Hiperplasia suprarrenal congénita no-clásica: avances en la detección, diagnóstico, conducta y tratamiento
Merino PM.1,2 y Codner E.1
Nonclassic adrenal hyperplasia: State of the art
1Instituto de Investigación Materno Infantil (I.D.I.M.I.).
Facultad de Medicina, Universidad de Chile.
2Ginecología de la Adolescencia. Unidad de Ginecología,
Clínica Las Condes. Santiago, Chile.
Correspondencia a:
Dra. Ethel Codner D.
Profesor Asociado Instituto de Investigación
Materno Infantil (I.D.I.M.I.).
Escuela de Medicina, Universidad de Chile, Casilla
226-3.
Santiago, Chile.
Teléfono: 562-9770865
FAX: 562-4247240
E-mail: ecodner@med.uchile.cl
Recibido: 16 de Noviembre de 2009
Aceptado: 14 de Diciembre de 2009
Nonclassical adrenal hyperplasia (NC-CAH) is caused by a deficiency in the activity of the 21-hydroxylase enzyme and is the most common autosomal recessive disorder. The clinical features of the disease are highly variable, and therefore the diagnosis may be overseen. The disorder is characterized by hyperandrogenism of adrenal origin that may become evident during childhood, adolescence or adulthood. The symptoms vary from premature pubarche, menstrual disturbances, hirsutism and virilization to those cases without any clinical evidence of the disease, as described in the cryptic form. The diagnostic approach includes an initial measurement of plasmatic 17OH-progesterone (17OHP) and androgen levels, and an ACTH test in those with elevated baseline 17OHP. The definitive diagnosis of this entity is performed with the documentation of abnormalities in both alleles of the CYP21A2 gene. This paper reviews the clinical, molecular and treatment of patients with NC-CAH.
Key words: Nonclassical congenital adrenal hyperplasia, adrenal hyperandrogenism,
deficiencia de 21-hidroxilasa, pubarquia prematura, Síndrome de Ovario Poliquístico.
La hiperplasia suprarrenal congénita (HSC) corresponde a un grupo de trastornos autosómicos recesivos secundarios a defectos en las enzimas que participan en la biosíntesis del cortisol1. El cuadro clásico de HSC, que corresponde a la forma de mayor gravedad, se manifiesta en los primeros años de vida y es causado por mutaciones que determinan un grave compromiso funcional de las enzimas esteroidogénicas. La gran mayoría de los casos de HSC clásica son causados por deficiencia de la 21hidroxilasa (21OH) secundaria a mutaciones en su gen CYP21A2. La deficiencia de 21OH produce un bloqueo en la conversión de 17OH-progesterona (17OHP) a 11-deoxicortisol y en la conversión de progesterona a deoxicorticosterona, afectando la síntesis de cortisol y aldosterona, respectivamente. La 21OH no participa en las vías que sintetizan andrógenos, por lo que las mutaciones de ésta se acompañan de mayor producción de andrógenos adrenales por metabolismo de los compuestos que se acumulan previo al bloqueo2.
El cuadro no clásico de HSC (HSC-NC) se caracteriza por hiperandrogenismo de origen adrenal que se manifiesta durante la niñez o en el período de la adolescencia y adultez. Aunque existen comunicaciones de formas no clásicas por defectos de las otras enzimas, la mayoría de los casos se deben a alteraciones del CYP21A2. Las alteraciones genéticas que causan las formas no clásicas afectan en menor grado la actividad de la enzima en comparación con las formas clásicas, y no afectan en forma significativa la secreción de cortisol o mineralocorticoides. Esto determina que el aumento en la producción de andrógenos sea insuficiente para producir virilización de los genitales in utero o durante los primeros años de vida. El presente artículo describe el diagnóstico y manejo de la HSC-NC por deficiencia de 21OH.
Epidemiología
La HSC-NC es la enfermedad más frecuente con herencia autosómica recesiva. Su incidencia se estima en 1 en 1.000 individuos en población general, y en 10 a 20 y 30 a 40 en 1.000 en población hispánica y judíos Ashkenazi, respectivamente3,4. Se estima que 1 de cada 60 individuos en población blanca es portador de una mutación del CYP21A2, la que puede ser tan común como 1 en 20 a 1 en 3 individuos en población judía5,6.
Historia
La historia de la HSC-NC se remonta a los años 1960 cuando Jayle describe por primera vez un caso de virilización postpuberal secundario a un defecto en la hidroxilación del carbono 217. A partir de entonces se describieron casos de pacientes con hirsutismo, virilización e infertilidad con el antecedente de pubertad normal y características sexuales secundarias femeninas normales. A estas pacientes se las rotuló como formas leves, atenuadas, de aparición tardía o adquiridas de HSC8. Recién en los años 80 se comenzó a utilizar el concepto de HSC no clásica para designar a este cuadro clínico y este es el modo con el que se prefiere denominar actualmente a esta entidad9.
Genética molecular
Las enzimas involucradas en la esteroidogénesis están asociadas al citocromo P450, motivo por el cual se denominan CYP. La enzima 21-hidroxilasa es una proteína de 495 aminoácidos que se localiza en los microsomas de las células. Esta enzima está codificada por el gen CYP21A2 que se encuentra localizado entre las moléculas de HLA en el cromosoma 6p21.3, y tiene 10 exones y 9 intrones. Cercano al CYP21A2, aproximadamente 30kb arriba de este gen, se encuentra un pseudogen conocido como CYP21A1 o CYP21P, que no codifica para proteína activa. Este gen es idéntico en un 98% al CYP21A210,11.
La gran mayoría de los casos de HSC se deben a recombinación de ambos genes durante la meiosis, proceso conocido como "conversión génica", produciendo una proteína truncada y/o con menos actividad enzimática. El grado de compromiso de la actividad de la 21OH determina que existan genotipos con pérdida muy acentuada de la función enzimática y otras formas más leves. La presencia de dos alelos graves determina una HSC clásica, pero la existencia de al menos un alelo leve es compatible con la forma no clásica (Tabla 1).
Las alteraciones genéticas causantes de HSC-NC se describen en la Tabla 2 y corresponden a conversiones génicas y un pequeño porcentaje a mutaciones de novo. Los pacientes pueden tener dos mutaciones leves, pero un 66 a 75% de ellos corresponden a heterocigotos compuestos, en que un alelo puede tener una alteración que afecte en gran magnitud la actividad de la enzima, lo que tiene implicancias obstétricas12,13. La gran mayoría de HSC-NC están causadas por las mutaciones V281L y P30L.
Mutaciones del CYP21A2 que determinan la hiperplasia suprarrenal congénita no clásica, localización, y actividad enzimática.
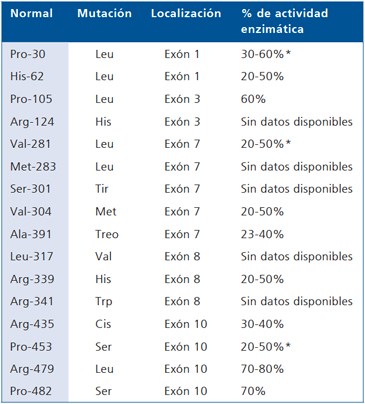 *Este estudio está disponible en Chile, y es realizado por análisis
por PCR alelo-específica en la Pontificia Universidad Católica. Incluye
el estudio de las siguientes anormalidades genéticas asociadas a
Hiperplasia Suprarrenal Congénita No-Clásica: Pro30leu, Val281leu,
Pro453Ser, y las siguientes alteraciones asociadas a las formas Clásicas:
deleción o LGC (macro conversión), G110+8nt, ILe172asn,
ClusterE6, Leu306+1nt, Gln318stop, Arg356Trp.
*Este estudio está disponible en Chile, y es realizado por análisis
por PCR alelo-específica en la Pontificia Universidad Católica. Incluye
el estudio de las siguientes anormalidades genéticas asociadas a
Hiperplasia Suprarrenal Congénita No-Clásica: Pro30leu, Val281leu,
Pro453Ser, y las siguientes alteraciones asociadas a las formas Clásicas:
deleción o LGC (macro conversión), G110+8nt, ILe172asn,
ClusterE6, Leu306+1nt, Gln318stop, Arg356Trp.
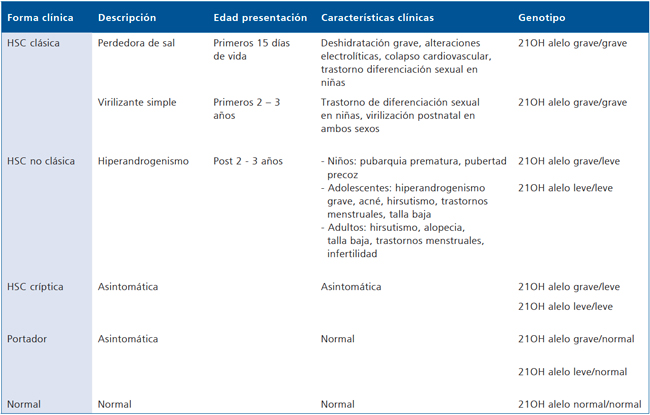
Tabla 2. Formas clínicas de la hiperplasia suprarrenal congénita. Características fenotípicas y genotipo
Manifestaciones clínicas
Existen cuatro formas clínicas de HSC que se definen por la edad de diagnóstico y la gravedad clínica (Tabla 1)10,14.
La HSC clásica tiene dos formas de presentación. La HSC clásica perdedora de sal se manifiesta en el período neonatal y se caracteriza por trastornos en la diferenciación sexual y un cuadro de deshidratación grave con hiponatremia e hiperkalemia. La forma HSC clásica virilizante simple se manifiesta por virilización al nacimiento y/o los 2 primeros años de vida sin pérdida de sal15.
La presentación clínica de la HSC-NC es la de un hiperandrogenismo adrenal que aparece después de los 2 a 3 años de vida y puede manifestarse en la niñez, pubertad o durante la vida adulta. En algunos casos existe el trastorno molecular y bioquímico, pero sin signos clínicos, forma conocida como HSC críptica. La serie más grande de nuestro país, que incluyó 57 pacientes, fue publicada recientemente por Martínez-Aguayo et al16.
Durante la niñez la principal manifestación de la HSC-NC es la pubarquia prematura, que es la aparición de vello púbico antes de los 8 años en las niñas y antes de los 9 años en los niños. La incidencia de HSC-NC en niños con pubarquia prematura oscila entre 4 y 20%17-18. Un estudio reciente en 238 niños y niñas con pubarquia precoz demostró una incidencia de HSC-NC18 de 4%. Otras manifestaciones clínicas durante la etapa escolar son la talla alta con avance de la edad ósea, clítoromegalia en la niña o crecimiento peneano con testículos pequeños en el niño. En ambos sexos, el aumento de los andrógenos adrenales puede acelerar la maduración del eje gonadal y desencadenar una pubertad precoz.
Durante la pubertad es más frecuente realizar el diagnóstico de HSC-NC en mujeres que en hombres, debido a la preocupación que produce el hiperandrogenismo en las adolescentes. El motivo de consulta principal es el hirsutismo, que está presente en la gran mayoría de las pacientes y que puede alcanzar gran magnitud19,20. Otros signos frecuentes son la amenorrea primaria u oligomenorrea (60%) y el acné (33%)21,22. La menarquia puede ser normal o tardía, sin embargo debe tenerse presente que ciclos menstruales regulares no descartan esta patología23. En los adolescentes varones el cuadro es difícil de diagnosticar, ya que los signos del exceso de andrógenos se confunden a menudo con la pubertad fisiológica. El cuadro debe ser sospechado en pacientes con acné marcado, pubertad precoz o rápidamente progresiva y un estirón puberal temprano con talla baja final.
Durante la adultez, semejante a lo que ocurre durante la pubertad, es más frecuente realizar el diagnóstico de HSC-NC en mujeres que consultan por hiperandrogenismo. La clínica puede ser similar a un Síndrome de Ovario Poliquístico (SOP) o a un hiperandrogenismo grave secundario a un tumor productor de andrógenos. Las pacientes heterocigotas compuestas, portadoras de una mutación grave, pueden tener síntomas de hiperandrogenismo más marcados13. La infertilidad es un hallazgo clínico que se puede observar en 15 a 30% de las mujeres adultas con HSC-NC, y está dada principalmente por la anovulación y la mayor frecuencia de abortos espontáneos21,22-24. El cuadro clínico de HSCNC en el hombre adulto es sutil, y a menudo se piensa en el diagnóstico a raíz de un caso índice en la familia. Las alteraciones en la fertilidad, observadas en los individuos con la forma clásica, se dan en menor grado en pacientes con HSC-NC y a menudo son reversibles25,26. Pinkas estudió la prevalencia de HSC-NC en 222 hombres con un espermiograma alterado y la comparó con la de 262 controles, no encontrando alteraciones en la esteroidogénesis adrenal en el grupo enfermo. Estos hallazgos no apoyan la medición rutinaria de 17OHP en la evaluación de la pareja infértil por causa masculina27.
La forma críptica, que no presenta signos clínicos, fue descrita por Levine al estudiar 8 familias de pacientes con HSC clásica. Él describe un grupo de pacientes supuestamente afectados que no manifestaban signos de hiperandrogenismo, a pesar de presentar niveles elevados de 17OHP28. Las hipótesis para tratar de explicar este fenómeno se han basado en una diferente respuesta de los tejidos periféricos a los andrógenos, ya sea por diferencias en el número de repeticiones CAG del receptor de testosterona o por cambios en la actividad de la P450 oxidoreductasa. Esta enzima parece ser el modulador principal de la expresión fenotípica de la deficiencia de 21-hidroxilasa, aunque algunos grupos cuestionan su real implicancia11,29. Otros grupos proponen la influencia del hiperinsulinismo en la producción ovárica de andrógenos, lo que discutiremos más adelante30,31.
Laboratorio
El diagnóstico de HSC-NC se basa fundamentalmente en la determinación de las concentraciones plasmáticas de 17OHP, basales y estimuladas con ACTH, y se certifica con el estudio molecular. La determinación de los niveles de mineralocorticoides, actividad de renina plasmática y cortisol no ayudan en la práctica clínica en el diagnóstico, ni en la evaluación a largo plazo del cuadro.
Tamizaje con 17OHP basal
La 17OHP basal es el precursor hormonal que se
acumula por la deficiencia de 21OH, y su medición es
el examen de laboratorio utilizado para tamizaje de la
enfermedad. La elevación de 17OHP puede ser de diferente
magnitud según el grado de compromiso de la actividad de
la enzima, existiendo superposición entre sujetos normales
y heterocigotos portadores de un alelo enfermo, así como
entre heterocigotos con un alelo afectado con sujetos con
ambos alelos comprometidos, a diferencia de lo que se ve
en las formas de aparición tardía que presentan niveles
menores que los de la forma clásica. Se han construido
nomogramas de referencia con los valores de 17OHP
basales y postestímulo con ACTH que son utilizados en el
diagnóstico de HSC (Figura 1)6,15.
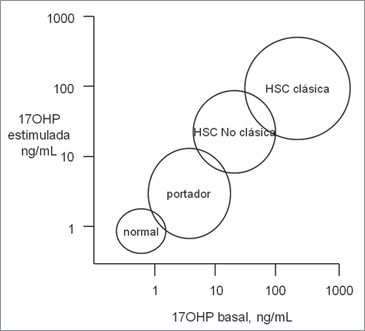 Figura 1. Nomograma con los valores de 17OHP basales y post estímulo
con 0,25 mg de ACTH en sujetos con y sin hiperplasia suprarrenal
congénita. Modificado de referencia 15.
Figura 1. Nomograma con los valores de 17OHP basales y post estímulo
con 0,25 mg de ACTH en sujetos con y sin hiperplasia suprarrenal
congénita. Modificado de referencia 15. Con la disponibilidad del estudio molecular para definir qué pacientes tienen ambos alelos alterados, se han buscado los puntos de corte que separen a los sujetos con cero, uno o dos alelos comprometidos. Dewailly, el año 1986, hace un análisis de 20 pacientes, donde sugiere que un valor basal de 5 ng/mL sería diagnóstico de HSC-NC y no requeriría evaluaciones funcionales adicionales32. Posteriormente, el año 1989, Azziz y Zacur publican un estudio de 164 mujeres con hiperandrogenismo y las comparan con 21 mujeres controles eumenorreicas y sin manifestaciones de exceso de andrógenos. Este estudio señala que niveles basales de 17OHP < 2 ng/mL descartarían la presencia de una HSCNC, y que valores entre 2 y 4 ng/mL pueden corresponder a sujetos normales, heterocigotos o con HSC-NC33. Los valores basales de 17OHP < 3 ng/mL son infrecuentes en estas pacientes19.
El valor del punto de corte de 17OHP que efectivamente sugiere la presencia de HSC-NC es especialmente importante en el caso de las pacientes con SOP, ya que éstas presentan un trastorno en la esteroidogénesis ovárica, que se ve reflejada en niveles más elevados de 17OHP, tanto basales como post estímulo con análogos de GnRH34,35. Carmina, en su estudio sobre 950 mujeres hiperandrogénicas describe valores basales de 17OHP de 1,4 ± 0,8 ng/mL en el grupo SOP, cifra superior y estadísticamente significativa respecto a los niveles de las mujeres con hirsutismo idiopático36. El estudio de Pall describe que un 20% de las mujeres con SOP tienen valores de 17OHP entre 2 y 3 ng/mL, lo que se observa principalmente en las SOP obesas19.
La determinación de 17OHP tiene diversas dificultades, por lo que debe ser realizada tomando las siguientes precauciones: efectuarla en un laboratorio de excelencia cuya técnica no tenga reacción cruzada con otros esteroides, obtención de la muestra en horario matinal dado el ritmo circadiano de la secreción de ACTH, en ayunas, sin tratamiento previo con corticoides y en fase folicular del ciclo para evitar la confusión con la producción de 17OHP por el cuerpo lúteo. Si el valor de 17OHP se encuentra persistentemente entre 3 y 4 ng/mL, se plantea que se trata de un individuo heterocigoto para mutación del CYP21A2 o se está ante un paciente con hiperandrogenismo ovárico o adrenal, por lo que sugerimos realizar prueba de estimulación con ACTH. El diagnóstico confirmatorio se realiza con el estudio molecular y es apoyado con la prueba de ACTH.
Prueba de estimulación con ACTH para medición
de 17OHP
La prueba de ACTH busca aclarar el diagnóstico en aquellos pacientes con niveles basales dudosos de 17OHP, y consiste en la administración iv de 25 ug de ACTH (Synacten®) con medición de 17OHP basal y a los 60 minutos. En forma semejante a lo que ocurre con los concentraciones basales de 17OHP existe un aumento progresivo en los niveles estimulados de este esteroide, desde los sujetos normales hasta los pacientes con HSC clásica (Figura 1). Se ha propuesto que valores estimulados < 10 ng/mL corresponden a sujetos sanos, entre 10 y 15 ng/mL a heterocigotos y > 15 ng/ml a pacientes con HSC-NC6,32. Bachega y cols, estudiaron la relación entre la gravedad de la mutación, las características clínicas y los niveles de 17OHP y demostraron que la respuesta a la prueba de ACTH es altamente variable y no se relaciona con el grado de déficit enzimático37.
Niveles circulantes de andrógenos
Se ha intentado utilizar los niveles de andrógenos séricos como elemento para diferenciar la HSC-NC del SOP o de una pubarquia prematura aislada. En el caso del SOP, algunos estudios no encuentran diferencias entre los niveles de testosterona y DHEAS respecto al grupo con HSC-NC, pero otros han descrito mayores niveles de estos esteroides en este último grupo12,33,36. La androstenediona se postula como un marcador efectivo que permitiría diferenciar estos dos grupos19. En la pubarquia prematura, el grupo con HSCNC presentaría niveles de androstenediona y testosterona superiores al grupo sin HSC18.
Estudio genético molecular de CYP21A2
El diagnóstico definitivo se realiza en base al estudio molecular, y se debe efectuar en todos los pacientes con estudio hormonal sugerente de HSC-NC. El estudio de las mutaciones del CYP21A2 se puede realizar por PCR alelo-específica o por secuenciación del gen. En Chile, el laboratorio de la P. Universidad Católica realiza el estudio por reacción de polimerasa en cadena alelo-específica para el estudio de 11 mutaciones, de las cuáles sólo tres son alelos leves asociados a HSC-NC (Tabla 1: Val281Leu, Pro453Ser, Pro30Leu)38. Este estudio detecta el 75-80% de las alteraciones genéticas causantes de HSC, por lo que en una proporción de pacientes se debe recurrir a la secuenciación del gen para aclarar el trastorno genético. La secuenciación del CYP21A2 se encuentra disponible en forma comercial en el extranjero (www.athenadiagnostics. com) y lo estará próximamente en nuestro país.
Complicaciones potenciales
Insuficiencia adrenal secundaria a tratamiento crónico con glucocorticoides
La insuficiencia suprarrenal es poco frecuente en pacientes con HSC-NC, a diferencia de la forma clásica en la que existe una alteración en la síntesis del cortisol. Sin embargo, esta complicación puede ocurrir si el paciente recibe dosis supra-fisiológicas de glucocorticoides, por lo que se recomienda que los pacientes con HSC-NC en tratamiento esteroidal, porten una credencial o brazalete que alerte sobre esta condición39.
Resistencia a la insulina y consecuencias
metabólicas
Existen hipótesis que sugieren un rol deletéreo de los andrógenos sobre la acción de la insulina y de ésta sobre la síntesis de andrógenos. Las pacientes con HSC-NC presentan niveles de insulina basal y postcarga más altos, y menor sensibilidad a la insulina en comparación con el grupo control30. Pall et al, no confirma las observaciones realizadas por el estudio anterior, mostrando valores comparables a los controles en glicemia e insulinemia basal y HOMA-IR19.
Disfunción ovárica
La HSC-NC se asocia con trastornos del ciclo menstrual, que aparecen en más de la mitad de las pacientes. Además, se puede observar una alteración en la secreción de las gonadotropinas, con una relación LH/FSH > 2 en el 9% de pacientes HSC-NC19.
El hiperandrogenismo ovárico funcional o hiperandrogenismo de causa ovárica, que se diagnostica en base a una mayor producción de 17OHP a nivel del ovario ante un estímulo con análogos de GnRH, se ha descrito como una de las complicaciones de las pacientes con HSC clásica, pero no está clara esta alteración en HSC-NC40.
Infertilidad
La infertilidad en pacientes con HSC-NC varía de un 16 a 30%, la cual en gran medida está determinada por la anovulación crónica que acompaña a la mitad de las pacientes21. Un estudio con más de 203 embarazos de mujeres con HSC-NC describe un 19,2% de abortos espontáneos, 0,5% de embarazos ectópicos y 1% de mortinatos. El número de abortos espontáneos es significativamente mayor antes del diagnóstico de HSC-NC (25,4%) que después de él (6,2%)41. Este resultado concuerda con reportes previos que describen un 33% de abortos en el grupo de pacientes no tratadas42.
Tratamiento
Hiperandrogenismo
El tratamiento estándar de la HSC-NC es el propio de los pacientes sintomáticos con glucocorticoides. Recientemente se publicó un consenso internacional que sugiere el tratamiento sólo en niños sintomáticos, con cortisol en dosis de 10-15 mg/m2/d, dividida en 3 dosis43. En adolescentes con crecimiento completo y en adultos, el tratamiento puede ser realizado con dexametasona en dosis nocturna de 0,25-0,5 mg. En mujeres adultas es posible cambiar el tratamiento corticoidal por un antiandrógeno potente como la ciproterona o espironolactona asociado a un anticonceptivo oral que contenga una progestina antiandrogénica4. No se recomienda el tratamiento con corticoides para las pacientes heterocigotas con un alelo sano y otro alterado. El control del tratamiento se realiza en base a los niveles de androstenediona y 17OHP.
Embarazo
El primer paso en la preparación del embarazo es la recuperación de la fertilidad con el tratamiento adecuado del hiperandrogenismo con glucocorticoides, el cual restablece la función ovulatoria41,42. En caso de ser necesario un inductor de ovulación como clomifeno o gonadotropinas, se deben tomar precauciones especiales respecto del síndrome de hiperestimulación ovárica.
Las pacientes con HSC-NC tienen un riesgo de 2,5% de tener un hijo con HSC clásica y de 14,8% de tener descendencia con HSC-NC41. El manejo obstétrico del embarazo en una mujer con HSC-NC depende del riesgo de virilización de un feto femenino, que ocurre sólo si padre y madre son portadores de alelos graves, por lo que se recomienda determinar el genotipo de ambos. Si el padre o madre no son portadores de un alelo grave, no existe posibilidad que el feto tenga una HSC clásica, por lo que la madre no debe ser tratada con dexametasona durante el embarazo, sino sólo con cortisol, medicamento que no atraviesa la barrera placentaria. La razón por la que la mujer con HSC-NC que tiene un cónyuge sano, no requiere tratamiento para prevenir la virilización del feto, se debe a que la aromatasa placentaria lo protegería de los niveles elevados de andrógenos de la madre. En el caso que ambos padres tengan un alelo grave, se puede realizar tratamiento prenatal para prevenir la virilización del feto femenino. Este tratamiento es aún experimental y debe ser efectuado con consentimiento y consiste en la administración de dexametasona en dosis de 20 μg/kg/d hasta el diagnóstico de sexo fetal45.
Conclusiones
Aunque la HSC-NC es el trastorno autosómico recesivo más frecuente, dada su amplia variabilidad fenotípica, sigue siendo poco diagnosticada en hombres y mujeres que consultan por hiperandrogenismo. Los pacientes portadores de un alelo enfermo, evento muy frecuente en la población, tienen valores levemente elevados de esteroides, pero no deben ser tratados con corticoides. Debemos tener presente que el diagnóstico definitivo se basa en la determinación del defecto genético, lo que además permitirá una adecuada consejería preconcepcional y el tratamiento de las complicaciones asociadas.
Referencias
- Speiser PW, White PC. 2003. Congenital adrenal hyperplasia. N Engl J Med 349: 776-788.
- Huerta R, Dewailly D, Decanter C, Knochenhauer ES, Boots LR, Azziz R. 2000. Adrenocortical hyperresponsivity to adrenocorticotropic hormone: a mechanism favoring the normal production of cortisol in 21-hydroxylase-deficient nonclassic adrenal hyperplasia. Fertil Steril 74: 329-334.
- White PC. 2001. Congenital adrenal hyperplasias. Best Pract Res Clin Endocrinol Metab; 15: 17-41.
- Knochenhauer ES, Cortet-Rudelli C, Cunnigham RD, Conway-Myers BA, Dewailly D, Azziz R. 1997. Carriers of 21-hydroxylase deficiency are not at increased risk for hyperandrogenism. J Clin Endocrinol Metab 82: 479-485.
- Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. 1985. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet 37: 650-667.
- New MI, Lorenzen F, Lerner AJ, Kohn B, Oberfield SE, Pollack Ms, et al. 1983. Genotyping steroid 21-hydroxylase deficiency: hormonal reference data. J Clin Endocrinol Metab 57: 320-326.
- Jayle MF, Weinmann SH, Baulieu EE, Vallin Y. 1958. [Isolated post-puberal virilism in the absence of hydroxylation of steroids in 21 C-21 position.]. Acta Endocrinol (Copenh) 29: 513-524.
- Blankstein J, Faiman C, Reyes Fi, Schroeder Ml, Winter Js. 1980. Adult-onset familial adrenal 21-hydroxylase deficiency. Am J Med 68: 441-448.
- Drucker S, New MI.1987. Nonclassic adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatr Clin North Am 34: 1067-1081.
- Robins T, Carlsson J, Sunnerhagen M, Wedell A, Persson B. 2006. Molecular model of human CYP21 based on mammalian CYP2C5: structural features correlate with clinical severity of mutations causing congenital adrenal hyperplasia. Mol Endocrinol 20: 2946-2964.
- Krone N, Arlt W. 2009. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab 23: 181-192.
- Azziz R, Dewailly D, Owerbach D. 1994. Clinical review 56: Nonclassic adrenal hyperplasia: current concepts. J Clin Endocrinol Metab 78: 810-815.
- Speiser PW, Knochenhauer ES, Dewailly D, Fruzzetti F, Marcondes JA, Azziz R. 2000. A multicenter study of women with nonclassical congenital adrenal hyperplasia: relationship between genotype and phenotype. Mol Genet Metab 71: 527-534.
- Levine LS. 2000. Congenital adrenal hyperplasia. Pediatr Rev 21: 159-170; quiz 171.
- White PC, Speiser PW. 2000. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 21: 245-291.
- Martínez-Aguayo A, Rumie H, Poggi M, García H, Mericq V, Arteaga E, et al. 2008. Hiperplasia suprarrenal no clásica, características clínicas y genéticas.Rev Chil Endocrinol Diabetes 2: 92-97.
- Siegel SF, Finegold DN, Urban MD, Mcvie R, Lee PA. 1992. Premature pubarche: etiological heterogeneity. J Clin Endocrinol Metab 74: 239-247.
- Armengaud JB, Charkaluk ML, Trivin C, Tardy V, Breart G, Brauner R, et al. 2009. Precocious pubarche: distinguishing lateonset congenital adrenal hyperplasia from premature adrenarche. J Clin Endocrinol Metab 94: 2835-2840.
- Pall M, Azziz R, Beires J, Pignatelli D. 2009. The phenotype of hirsute women: a comparison of polycystic ovary syndrome and 21-hydroxylase-deficient nonclassic adrenal hyperplasia. Fertil Steril, Epub DOI: 10.1016/j.fertnstert.2009.06.025
- Bidet M, Bellanne-Chantelot C, Galand-Portier MB, Tardy V, Billaud L, Laborde K, et al. 2009. Clinical and molecular characterization of a cohort of 161 unrelated women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency and 330 family members. J Clin Endocrinol Metab 94: 1570-1578.
- Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibáñez L, et al. 2000. 21-Hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder: a multicenter study. Am J Obstet Gynecol 183: 1468-1474.
- Speiser PW. 2009. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord 10: 77-82.
- Lin-Su K, Nimkarn S, New MI. 2008. Congenital adrenal hyperplasia in adolescents: diagnosis and management. Ann N Y Acad Sci 1135: 95-98.
- New MI. 2006. Extensive clinical experience: nonclassical
21-hydroxylase deficiency. J Clin Endocrinol Metab 91: 4205-
4214. - Kalachanis I, Rousso D, Kourtis A, Goutzioulis F, Makedos G, Panidis D. 2002. Reversible infertility, pharmaceutical and spontaneous, in a male with late onset congenital adrenal hyperplasia, due to 21-hydroxylase deficiency. Arch Androl 48: 37-41.
- Augarten A, Weissenberg R, Pariente C, Sack J. 1991. Reversible male infertility in late onset congenital adrenal hyperplasia. J Endocrinol Invest 14: 237-240.
- Pinkas H, Fuchs S, Klipper-Aurbach Y, Zvulunov A, Raanani H, Mimouni G, et al. 2009. Non-classical 21-hydroxylase deficiency: prevalence in males with unexplained abnormal sperm analysis. Fertil Steril, Epub DOI 10.1016/j.fertnstert.2008.12.037
- Levine LS, Dupont B, Lorenzen F, Pang S, Pollack M, Oberfield S, et al. 1980. Cryptic 21-hydroxylase deficiency in families of patients with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab 51: 1316-1324.
- Huang N, Agrawal V, Giacomini KM, Miller WL. 2008. Genetics of P450 oxidoreductase: sequence variation in 842 individuals of four ethnicities and activities of 15 missense mutations. Proc Natl Acad Sci USA 105: 1733-1738.
- Saygili F, Oge A, Yilmaz C. 2005. Hyperinsulinemia and insulin insensitivity in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency: the relationship between serum leptin levels and chronic hyperinsulinemia. Horm Res 63: 270-274.
- Speiser PW, Serrat J, New MI, Gertner JM. 1992. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab 75: 1421- 1424.
- Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. 1986. Clinical and biological phenotypes in late-onset 21-hydroxylase deficiency. J Clin Endocrinol Metab 63: 418-423.
- Azziz R, Zacur HA. 1989. 21-Hydroxylase deficiency in female hyperandrogenism: screening and diagnosis. J Clin Endocrinol Metab 69: 577-584.
- Barnes RB, Rosenfield RL, Burstein S, Ehrmann DA. 1989. Pituitary-ovarian responses to nafarelin testing in the polycystic ovary syndrome. N Engl J Med 320: 559-565.
- Rosenfield RL, Barnes RB, Cara JF, Lucky AW. 1990. Dysregulation of cytochrome P450c 17 alpha as the cause of polycystic ovarian syndrome. Fertil Steril 53: 785-791.
- Carmina E, Rosato F, Janni A, Rizzo M, Longo RA. 2006.
Extensive clinical experience: relative prevalence of different
androgen excess disorders in 950 women referred because of
clinical hyperandrogenism. J Clin Endocrinol Metab 91: 2-6.
- Bachega TA, Billerbeck AE, Marcondes JA, Madureira G, Arnhold IJ, Mendonca BB. 2000. Influence of different genotypes on 17-hydroxyprogesterone levels in patients with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol (Oxf) 52: 601-607.
- Fardella CE, Poggi H, Pineda P, Soto J, Torrealba I, Cattani A, et al. 1998. Salt-wasting congenital adrenal hyperplasia: detection of mutations in CYP21B gene in a Chilean population. J Clin Endocrinol Metab 83: 3357-3360.
- Bornstein SR. 2009. Predisposing factors for adrenal insufficiency. N Engl J Med 360: 2328-2339.
- Ghizzoni L, Virdis R, Vottero A, Cappa M, Street Me, Zampolli M, et al. 1996. Pituitary-ovarian responses to leuprolide acetate testing in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 81: 601-606.
- Moran C, Azziz R, Weintrob N, Witchel Sf, Rohmer V, Dewailly D, et al. 2006. Reproductive outcome of women with 21-hydroxylase-deficient nonclassic adrenal hyperplasia. J Clin Endocrinol Metab 91: 3451-3456.
- Feldman S, Billaud L, Thalabard Jc, Raux-Demay MC, Mowszowicz I, Kuttenn F, et al. 1992. Fertility in women with late-onset adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 74: 635-639.
- Consensus statement on 21-hydroxylase deficiency from the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. 2002. J Clin Endocrinol Metab 87: 4048-4053.
- Spritzer P, Billaud L, Thalabard Jc, Birman P, Mowszowicz I, Raux-Demay Mc, et al. 1990. Cyproterone acetate versus hydrocortisone treatment in late-onset adrenal hyperplasia. J Clin Endocrinol Metab 70: 642-646.
- Merino P, Bachega T, Cespedes P, Trejo L, Billerbeck AE, Codner E. 2007. Molecular study of CYP21A2 gene for prenatal diagnosis of congenital adrenal hyperplasia. Report of a family]. Rev Méd Chile 135: 1450-1455.