El tejido graso como modulador endocrino: Cambios hormonales asociados a la obesidad
René Baudrand B.1, Eugenio Arteaga U.1 y Manuel Moreno G.2
Adipose tissue as an endocrine modulator: Hormonal changes associated with obesity
1Departamento de Endocrinología.
2Programa de Obesidad, Departamento de Nutrición,
Diabetes y Metabolismo. Facultad de Medicina,
Pontificia Universidad Católica de Chile.
Santiago de Chile.
Correspondencia:
Dr. René Baudrand Biggs
Lira 85, 5to piso, Departamento de Endocrinología,
Facultad de Medicina,
Pontificia Universidad Católica de Chile.
Fax: 6385675
E-mail: rbaudrand@med.puc.cl
Adipose tissue not only stores fat, but secretes factors and hormones, which modify the regulation, metabolism and secretion of several other hormones. The objective of this review is to describe the hormonal disorders associated with increased adipose tissue, which acts as a modulator or disruptor of the endocrine physiology, with special reference to cortisol, androgens, growth hormone and thyroid axis, and discuss the implications for the management and treatment of these patients.
Key words: Adipose tissue; Hormones; Obesity.
Reproducido con la autorización de la Revista Médica de Chile (Rev Med Chile 2010; 138: 1294-1301).
La obesidad es una enfermedad crónica caracterizada por un aumento patológico de grasa corporal que se asocia a un mayor riesgo para la salud. Se produce por un balance calórico positivo, ya sea por elevado aporte calórico, reducción del gasto energético o combinación de ambos.
Actualmente, se considera a la obesidad la epidemia del siglo XXI, afectando a hombres y mujeres de todas las edades. En Chile, según la encuesta nacional de salud 2004, la prevalencia de obesidad en adultos es de 19% para los hombres y 25% para las mujeres, siendo más prevalente a mayor edad, en el sexo femenino y en el estrato socioeconómico bajo.
La obesidad, especialmente la de tipo central o androide, se asocia a una mayor prevalencia de patologías crónicas, tales como hipertensión arterial (HTA), diabetes mellitus tipo 2 (DM2) e insulino resistencia (IR), colelitiasis, hígado graso no alcohólico (HGNA), dislipidemia, cardiopatía coronaria, cáncer, enfermedades respiratorias, psiquiátricas y osteoarticulares, las cuales limitan las expectativas y calidad de vida, representando un serio problema para la salud pública mundial1.
En la obesidad, la mayoría de los casos son de origen multifactorial, reconociéndose factores genéticos, metabólicos, endocrinológicos y ambientales. Sólo 2-3% de los obesos tendría como causa alguna patología endocrinológica, entre las que destacan el hipotiroidismo (que produce leve alza de peso), síndrome de Cushing, hipogonadismo y lesiones hipotalámicas asociadas a hiperfagia2.
En contraste con la poca frecuencia de causas endocrinológicas causantes de obesidad, el exceso progresivo de tejido graso puede producir secundariamente alteraciones de la regulación, metabolización y secreción de diferentes hormonas. Estos últimos serían reversibles con la baja de peso, siendo el ejemplo clínico más importante el del hiperinsulinismo secundario a mayor resistencia a insulina inducida por el sobrepeso.
El objetivo de esta revisión es describir algunos trastornos hormonales que se han estudiado recientemente, los cuales son derivados del aumento del tejido graso, que actúa como un modulador o disruptor de la fisiología endocrina, con especial referencia al cortisol, los andrógenos, la hormona del crecimiento y eje tiroideo, y discutir sus implicancias en el manejo y tratamiento de estos pacientes.
El tejido adiposo como órgano endocrino
En los últimos años se ha comprobado que el tejido adiposo no sólo almacena lípidos, sino que secreta numerosas hormonas siendo el órgano endocrino de mayor tamaño del organismo, equivalente a 10 a 60% del peso total de un individuo según su composición corporal e IMC3. A diferencia de otras glándulas endocrinas, su masa es variable y puede aumentar o disminuir progresivamente de tamaño dependiendo de la edad, ingesta alimentaria, actividad física, programación perinatal y predisposición genética (Figura 1)4.
Se ha demostrado que diferentes células del tejido graso secretan numerosos factores o adipoquinas que participan en el metabolismo sistémico, la regulación del ciclo menstrual, la coagulación y resistencia vascular. Algunos de estos factores tienen efecto principalmente paracrino y otros tienen importantes efectos sistémicos describiéndose recientemente el eje hipotálamo-hipófisis-tejido adiposo5.
Destacan dentro de los factores secretados la leptina, que participa en la regulación del balance energético y la secreción de gonadotrofinas; el PAI-1, y el angiotensinógeno en la homeostasis vascular e hipertensión arterial; la resistina, adiponectina, TNF α y visfatina en promover o disminuir la insulino resistencia; IL-6, TNF α,y PPAR que participan en el proceso de microinflamación; y enzimas que participan en la regulación hormonal, activando o inactivando hormonas, como la aromatasa a los andrógenos y la 11β-hidroxiesteroide deshidrogenasa al cortisol (11β-HSD)4,6.
Obesidad y cortisol
El cortisol es uno de los esteroides más importantes en la especie humana ya que controla el metabolismo lipídico, proteico y glucídico. Además, es importante en el manejo hidroelectrolítico y por ende de la presión arterial7.
El aumento del cortisol o glucocorticoides en la sangre, se observa en el síndrome de Cushing, tanto endógeno o exógeno. Este hipercortisolismo genera un fenotipo caracterizado por obesidad centrípeta, diabetes o resistencia insulínica e hipertensión arterial, asemejándose mucho a las alteraciones descritas en la obesidad central y sus trastornos metabólicos asociados, que característicamente presenta niveles de cortisol plasmáticos normales pese a tener una tasa de secreción de cortisol aumentada18.
Dadas las semejanzas existentes entre el síndrome metabólico (SM) que incorpora la obesidad central, la hipertensión, la dislipidemia y la resistencia a insulina con el hipercortisolismo, se ha planteado que podría haber una alteración a nivel tisular en la regulación del cortisol9,10. El control local de la producción de glucocorticoides está a cargo de la enzima 11β-HSD1, la cual convierte la cortisona inactiva en cortisol, incrementando así la estimulación del receptor nuclear específico11. Esta enzima está ampliamente expresada en hígado y tejido adiposo visceral y subcutáneo, pero también en glándulas suprarrenales, ovario, placenta y células dendríticas12.
La sobreexpresión de la 11β-HSD1 en animales transgénicos resulta en hígado graso y dislipidemia, HTA, obesidad y resistencia a la insulina13. Por el contrario, los animales "knock out" para 11β-HSD1 presentan disminución de la grasa visceral, menores niveles de TNF-α y mayores concentraciones de adiponectina14.
Estudios en humanos han demostrado que los obesos expresan más la 11β-HSD1 en tejido graso en comparación con sujetos no obesos, observándose que el cortisol generado localmente repercutiría en el grado de diferenciación de los adipocitos15. Se establecería entonces un círculo vicioso en los obesos debido a que la mayor expresión de 11β-HSD1 visceral promueve la acumulación de más tejido adiposo metabólicamente activo16.
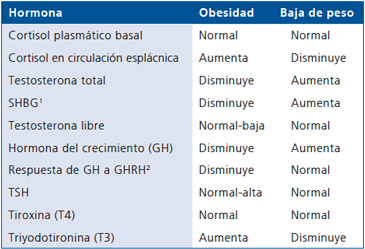
Una importante proporción del cortisol que circula en territorio esplácnico sería generado por la actividad de la 11β-HSD1 en hígado y tejido graso visceral, lo que generaría un hipercortisolismo a nivel pancreático y portal que explicaría algunas de las similitudes metabólicas entre la obesidad y el síndrome de Cushing (Figura 1). Una mayor producción esplácnica de cortisol en la obesidad no se reflejaría a nivel sistémico ya que es metabolizado principalmente a nivel hepático a sus tetrahidrometabolitos inactivos y podría explicar las diferencias fenotípicas con el síndrome de Cushing, como la ausencia de atrofia muscular proximal, equimosis fácil y estrías violáceas (Tabla 1)9.
Debido a estos nuevos hallazgos, algunos autores consideran a la obesidad como un "síndrome de Cushing visceral"8, planteando que el hipercortisolismo generado a nivel local tendría un papel fundamental en la perpetuación de la obesidad y en la patogenia de muchos trastornos metabólicos asociados, como recientemente fue publicado por nuestro grupo17. Esto abre un interesante campo de investigación para un futuro tratamiento farmacológico de la obesidad y del SM utilizando inhibidores específicos de la enzima 11β-HSD118.
Obesidad y andrógenos
La distribución de la grasa corporal sigue un patrón dimórfico19. En los hombres la acumulación de tejido graso es de predominio central o abdominal, lo que se ha denominado obesidad de tipo androide. En la mujer en cambio, se describe la obesidad de tipo periférica o ginecoide, con una mayor acumulación de grasa en la región glúteo-femoral20.
La grasa visceral presente en la obesidad central, que incluye el tejido omental, mesentérico y retroperitoneal, es reconocida en la actualidad como la manifestación de mayor prevalencia del SM, identificado como un perímetro abdominal aumentado y es la que más se asocia a diabetes tipo 2 y factores de riesgo cardiovascular21.
La relación entre el hipogonadismo y la adiposidad es compleja. Parte de la información relevante proviene del estudio de obesos con hipopituitarismo donde la condición de hipogonadismo se asocia a cambios en la composición corporal, caracterizados por menor masa magra y aumento de la masa grasa20.
Pero más interesante aun es comprobar que el fenómeno inverso, es decir, que los sujetos obesos suelen tener más hipogonadismo. Así, se ha demostrado que la cantidad de tejido adiposo visceral se asocia inversamente a los niveles de testosterona plasmática20. Estudios epidemiológicos prospectivos muestran que tanto el hipogonadismo es predictor de un mayor riesgo de desarrollar SM y DM222, como que la presencia de SM es un factor de riesgo de desarrollar hipogonadismo (Figura 1)23.
Una explicación fisiopatológica de la disminución de andrógenos secundaria a obesidad sería que el tejido adiposo expresa numerosas enzimas capaces de modificar esteroides, como por ejemplo, la aromatasa que cataliza la conversión de testosterona a estradiol y de androstenediona a estrona24.
Nuevas investigaciones sugieren además que las enzimas de la familia aldoketoreductasa 1C son muy activas en el tejido graso visceral y subcutáneo y pueden inactivar la dihidrotestosterona (DHT), el andrógeno de mayor potencia, especialmente en adipocitos maduros25. Estos estudios apoyan la hipótesis de que a mayor grasa corporal total hay mayor conversión de andrógenos a estrógenos y mayor inactivación de DHT, lo que favorecería el desarrollo de hipogonadismo descrito en sujetos obesos.
Pese a que en obesidad central masculina se asocia con testosterona total baja, el grado de obesidad, medido por tomografía axial computada (TAC) o resonancia magnética (RM), se asocia inversamente con los niveles de globulina hepática transportadora de esteroides sexuales (SHBG), lo que es explicado al menos parcialmente por la resistencia a la insulina que disminuye la SHBG26,27. La coexistencia de valores bajos tanto de testosterona como de SHBG explicaría por qué la testosterona libre se mantiene en rango normalbajo y por qué sólo una fracción de los hombres obesos presentan síntomas o signos categóricos de hipogonadismo (Tabla 1)1.
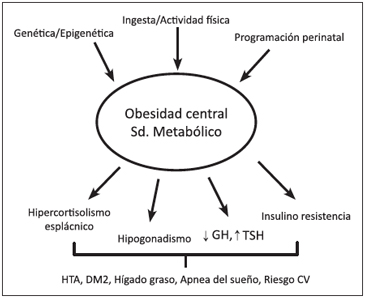
Figura 1. Patogenia y consecuencias clínicas de la obesidad central y el síndrome metabólico. GH: Hormona del crecimiento; TSH: Tirotropina; CV: cardiovascular.
Tabla 1. Cambios hormonales asociados a la obesidad y su evolución post baja de peso
 1. SHBG: globulina hepática transportadora de esteroides sexuales. 2. GHRH: factor liberador de hormona del crecimiento.
1. SHBG: globulina hepática transportadora de esteroides sexuales. 2. GHRH: factor liberador de hormona del crecimiento.
Respecto al tratamiento, un estudio realizado en hombres obesos sin hipogonadismo demostró que la administración de testosterona se asociaba a disminución del tejido adiposo visceral con mejoría de la insulino resistencia y el colesterol total, pero sin cambios significativos en grasa subcutánea o masa magra28.
En otros estudios, sin embargo, en pacientes hipogonádicos, que incluyen a sujetos con VIH e importante baja de peso, usuarios de glucocorticoides y en adultos mayores, se ha demostrado que la administración de testosterona induce también aumento de masa magra20,29,30. También se han realizado estudios aleatorizados en hombres y mujeres mayores de 60 años administrando dehidroepiandrosterona (DHEA) oral, lo que se asoció a disminución de grasa visceral y subcutánea, y mejoría de la insulina resistencia31.
Dada la información precedente, algunos autores han propuesto al hipogonadismo como un factor importante en el desarrollo y mantención de la obesidad central, SM, disfunción eréctil e infertilidad en población masculina adulta con sobrepeso32. Algunos de estos fenómenos pueden ser reversibles al bajar de peso, como se ha demostrado al menos en relación a disfunción eréctil e infertilidad, lo que apoya la hipótesis de que el tejido graso genera hipogonadismo secundario y refuerza la importancia de la baja de peso33.
Obesidad y hormona del crecimiento (GH)
La hormona del crecimiento (GH) tiene una conocida acción lipolítica, especialmente del tejido adiposo visceral (TAV), además de aumentar el gasto de energía y la masa magra.
Estudios en pacientes obesos han demostrado que tanto la GH basal como la secreción integrada de 24 horas están reducidas. La disminución de GH a mayor índice de masa corporal, con su consecuente menor lipólisis y menor gasto energético, se explicaría tanto por una disminución de la producción como por un aumento de la metabolización34. Se ha descrito también que los ácidos grasos libres circulantes, habitualmente elevados en obesos, inhiben la secreción de GH y podría ser una explicación fisiopatológica35.
Pese a que el ritmo circadiano de GH se mantiene en los obesos, los pulsos son de menor amplitud y con mayor intervalo. Además el "peak" nocturno de GH durante el sueño se reduce significativamente36. La patogenia de estos fenómenos, aunque no totalmente dilucidada, estaría relacionada a una disfunción del eje hipotálamo-hipofisiario o a un aumento de la acción inhibitoria de somatostatina1; situación que es reversible al bajar de peso (Tabla 1)37.
Con respecto al tratamiento con GH, un reciente meta-análisis de 24 estudios sugiere que el uso de GH recombinante en pacientes obesos también disminuye la adiposidad visceral y aumenta la masa magra significativamente, mejorando el perfil lipídico sin inducir pérdida de peso y con escasos efectos secundarios38.
Dado que las dosis utilizadas en muchos estudios son suprafisiológicas, consideramos necesario contar con estudios de mayor duración, que determinen la dosis mínima necesaria de GH para lograr estos objetivos y que exploren sus efectos sobre la morbi-mortalidad cardiovascular y la relación costo- efectividad.
Obesidad y hormonas tiroideas
Es una conducta ampliamente aceptada en la práctica clínica la evaluación de la función tiroidea en todo sujeto obeso. En estudios de tamizaje se ha descrito hipotiroidismo en 21% en el Programa de Obesidad de la Pontificia Universidad Católica de Chile39 y 25% de hipotiroidismo subclínico en obesos mórbidos previo a cirugía bariátrica40. Los estudios han dado lugar a resultados contradictorios y múltiples hipótesis, dado que pudiese existir un sesgo en el tamizaje y múltiples asociaciones metabólicas y hormonales.
Estudios en adultos obesos han demostrado que el exceso de peso se asocia a T4 normal y T3 aumentada por la sobrealimentación, asociado además a una elevación leve de TSH, y a un mayor volumen tiroideo que se correlaciona con la masa corporal magra, pero no con la masa grasa, cuyo mecanismo aún no está dilucidado (Tabla 1)41.
En oposición a la visión clínica tradicional de que una elevación de TSH o hipertirotropinemia implica algún grado de hipotiroidismo, otros estudios han propuesto que la hipertirotropinemia leve pudiese representar un mecanismo adaptativo a la obesidad y estar mediada por varios factores. Entre estos se describen un aumento de la leptina, que estimula la producción de pro-TRH, o un menor número de receptores de T3 en el hipotálamo que alteraría el sistema de retroalimentación. Aparentemente, la carencia de yodo o la autoinmunidad no serían determinantes en la frecuente elevación de TSH en obesos42.
Una nueva línea de investigación respecto a la patogénesis de las alteraciones tiroideas en la obesidad estaría relacionada con la acción de la insulina, la cual como factor de crecimiento participaría en la carcinogénesis de los tumores diferenciados de tiroides43. Recientemente, Rezzonico et al reportaron que mayores niveles de insulina se correlacionan con mayor volumen tiroideo y frecuencia de nódulos por ecografía, tanto en pacientes obesos como normopesos, lo que podría ser mediado por IGF-144. De esta forma se puede considerar que la glándula tiroides es otro órgano blanco impactado por el síndrome de resistencia a la insulina. Con respecto a la función tiroidea, se ha descrito en hombres obesos una directa correlación entre niveles de insulina y TSH, sobre todo con una insulinemia basal mayor a 21 uU/mL, efecto que se va atenuando con la edad45.
El aumento de TSH asociado a obesidad suele revertir después de la corrección del sobrepeso con cirugía bariátrica40; la excepción está dada por pacientes con marcadores de autoinmunidad tiroidea y aquellos con hipotiroidismo post-quirúrgico o post-actínico, los que deben considerarse como hipotiroideos genuinos46. Por otro lado, el uso de metformina reduce los niveles de TSH en mujeres diabéticas obesas con hipotiroidismo primario en terapia con levotiroxina, lo que apoya la hipótesis de que la hiperinsulinemia es la responsable del aumento de TSH, cuyo mecanismo aún no está bien dilucidado47.
Estos nuevos datos sugieren que el exceso de tejido graso produce una disfunción tiroidea reversible, lo que constituiría un nuevo beneficio de la corrección de la obesidad y pone una nota de cautela sobre el real significado de los hipotiroidismos subclínicos en estos sujetos.
Otros trastornos hormonales en pacientes obesos
Se ha descrito un aumento de la actividad del sistema renina-angiotensina-aldosterona en la obesidad. La aldosterona suele estar elevada en personas hipertensas con sobrepeso, mientras que la renina se mantiene por lo general dentro de la los niveles normales. Además, algunos autores han propuesto que existe una correlación positiva entre peso corporal y la relación aldosterona/actividad de renina plasmática1.
Estudios en humanos obesos han reportado un aumento de hormona paratiroidea (PTH) con calcemia normal. Esto corresponde a un hiperparatiroidismo secundario que podría explicarse por el déficit relativo de vitamina D por secuestro de 25-OH-hidroxivitamina D en el tejido graso48,49.
Conclusiones
Existen numerosas alteraciones endocrinas asociadas a la obesidad y el SM. Algunas de estas anomalías son consideradas como factores causales para el desarrollo del exceso de tejido adiposo, mientras que otras son inducidas por la obesidad y por lo general son corregibles con la baja de peso (Tabla 1).
La presencia de múltiples trastornos endocrinos asociados a la obesidad refuerza el nuevo concepto respecto a que el tejido graso es un órgano endocrino dinámico, muy activo en secreción de factores y adipoquinas y modificación enzimática de hormonas circulantes. Todo lo anterior explica gran parte de las alteraciones endocrinológicas, lo que ha permitido conocer más la fisiopatología de la obesidad y ha aumentado el conocimiento de los beneficios que se reconocen a la baja de de peso (Figura 1).
Dada la evidencia actual, es de vital importancia reconocer y buscar dirigidamente trastornos endocrinos prevalentes en la obesidad como el hipotiroidismo subclínico (o hipertirotropinemia), el hipogonadismo y el déficit de GH, cuya sustitución hormonal en casos seleccionados puede disminuir la morbilidad y las alteraciones bioquímicas y antropométricas asociadas.
Para el futuro quedan aún interrogantes pendientes respecto al impacto clínico en el largo plazo de la terapia con andrógenos o GH en pacientes obesos o el rol del control de la insulino resistencia en el hipotiroidismo subclínico. Existe también una activa investigación respecto al papel de la producción local de cortisol a nivel visceral y hepático y su relación con la obesidad, la hipertensión arterial, la dislipidemia y la insulino resistencia, como también su papel en la perpetuación del sobrepeso, lo que abre nuevas perspectivas de tratamientos farmacológicos específicos.
Referencias
- Kokkoris P, Pi-Sunyer FX. Obesity and endocrine disease. Endocrinol Metab Clin North Am 2003; 32: 895-914.
- Weaver JU. Classical endocrine diseases causing obesity. Front Horm Res 2008; 36: 212-28.
- Rocha VZ, Libby P. The multiple facets of the fat tissue. Thyroid 2008; 18: 175-83.
- Trujillo ME, Scherer PE. Adipose tissue-derived factors: impact on health and disease. Endocr Rev 2006; 27: 762-78.
- Schaffler A, Scholmerich J, Buechler C. The role of 'adipotropins' and the clinical importance of a potential hypothalamic-pituitary-adipose axis. Nat Clin Pract Endocrinol Metab 2006; 2: 374-83.
- Ronti T, Lupattelli G, Mannarino E. The endocrine function of adipose tissue: an update. Clin Endocrinol (Oxf) 2006; 64: 355-65.
- Frey FJ, Odermatt A, Frey BM. Glucocorticoid-mediated mineralocorticoid receptor activation and hypertension. Curr Opin Nephrol Hypertens 2004; 13: 451-8.
- Bujalska IJ, Kumar S, Stewart PM. Does central obesity reflect "Cushing's disease of the omentum"? Lancet 1997 26; 349 (9060): 1210-3.
- Basu R, Singh RJ, Basu A, Chittilapilly EG, Johnson CM, Toffolo G, et al. Splanchnic cortisol production occurs in humans: evidence for conversion of cortisone to cortisol via the 11-beta hydroxysteroid dehydrogenase (11beta-hsd) type 1 pathway. Diabetes 2004; 53: 2051-9.
- Iwasaki Y, Takayasu S, Nishiyama M, Tsugita M, Taguchi T, Asai M, et al. Is the metabolic syndrome an intracellular Cushing state? Effects of multiple humoral factors on the transcriptional activity of the hepatic glucocorticoid-activating enzyme (11beta-hydroxysteroid dehydrogenase type 1) gene. Mol Cell Endocrinol 2008 26; 285(1-2): 10-8.
- Draper N, Stewart PM. 11beta-hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J Endocrinol 2005; 186: 251-71.
- Freeman L, Hewison M, Hughes SV, Evans KN, Hardie D, Means TK, et al. Expression of 11beta-hydroxysteroid dehydrogenase type 1 permits regulation of glucocorticoid bioavailability by human dendritic cells. Blood 2005 15; 106: 2042-9.
- Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001; 7: 294 (5549): 2166-70.
- Morton NM, Paterson JM, Masuzaki H, Holmes MC, Staels B, Fievet C, et al. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11 beta-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 2004; 53: 931-8.
- Mariniello B, Ronconi V, Rilli S, Bernante P, Boscaro M, Mantero F, et al. Adipose tissue 11beta-hydroxysteroid dehydrogenase type 1 expression in obesity and Cushing's syndrome. Eur J Endocrinol 2006; 155: 435-41.
- Wolf G. Glucocorticoids in adipocytes stimulate visceral obesity. Nutr Rev 2002; 60 (5 Pt 1): 148-51.
- Baudrand R, Carvajal CA, Riquelme A, Morales M, Solís N, Pizarro M, et al. Overexpression of 11beta-Hydroxysteroid Dehydrogenase Type 1 in Hepatic and Visceral Adipose Tissue is Associated with Metabolic Disorders in Morbidly Obese Patients. Obes Surg 2010; 20: 77-83.
- Tomlinson JW, Sherlock M, Hughes B, Hughes SV, Kilvington F, Bartlett W, et al. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 activity in vivo limits glucocorticoid exposure to human adipose tissue and decreases lipolysis. J Clin Endocrinol Metab 2007; 92: 857-64.
- Blouin K, Boivin A, Tchernof A. Androgens and body fat distribution. J Steroid Biochem Mol Biol 2008; 108 (3-5): 272-80.
- Miller KK. Androgen deficiency: effects on body composition. Pituitary 2009; 12: 116-24.
- Vertemati M, Goffredi M, Moscheni C, Callegari S, Vizzotto L. Human visceral fat in different anthropometric patterns and in diabetes: a morphometric study. Anal Quant Cytol Histol 2008; 30: 39-46.
- Oh JY, Barrett-Connor E, Wedick NM, Wingard DL. Endogenous sex hormones and the development of type 2 diabetes in older men and women: the Rancho Bernardo study. Diabetes Care 2002; 25: 55-60.
- Laaksonen DE, Niskanen L, Punnonen K, Nyyssonen K, Tuomainen TP, Valkonen VP, et al. The metabolic syndrome and smoking in relation to hypogonadism in middle-aged men: a prospective cohort study. J Clin Endocrinol Metab 2005; 90: 712-9.
- Zumoff B. Hormonal abnormalities in obesity. Acta Med Scand Suppl 1988; 723: 153-60.
- Blouin K, Veilleux A, Luu-The V, Tchernof A. Androgen metabolism in adipose tissue: Recent advances. Mol Cell Endocrinol 2009; 25; 301 (1-2): 97-103.
- Khaw KT, Barrett-Connor E. Lower endogenous androgens predict central adiposity in men. Ann Epidemiol 1992; 2: 675-82.
- Gapstur SM, Gann PH, Kopp P, Colangelo L, Longcope C, Liu K. Serum androgen concentrations in young men: a longitudinal analysis of associations with age, obesity, and race. The CARDIA male hormone study. Cancer Epidemiol Biomarkers Prev 2002; 11 (10 Pt 1): 1041-7.
- Marin P, Holmang S, Jonsson L, Sjostrom L, Kvist H, Holm G, et al. The effects of testosterone treatment on body composition and metabolism in middle-aged obese men. Int J Obes Relat Metab Disord 1992; 16: 991-7.
- Stanworth RD, Jones TH. Testosterone for the aging male; current evidence and recommended practice. Clin Interv Aging 2008; 3: 25-44.
- Kapoor D, Jones TH. Androgen deficiency as a predictor of metabolic syndrome in aging men: an opportunity for intervention? Drugs Aging 2008; 25: 357-69.
- Villareal DT, Holloszy JO. Effect of DHEA on abdominal fat and insulin action in elderly women and men: a randomized controlled trial. JAMA 2004; 10; 292: 2243-8.
- Goulis DG, Tarlatzis BC. Metabolic syndrome and reproduction: I. testicular function. Gynecol Endocrinol 2008; 24: 33-9.
- Pasquali R, Patton L, Gambineri A. Obesity and infertility. Curr Opin Endocrinol Diabetes Obes 2007; 14: 482-7.
- Weltman A, Weltman JY, Hartman ML, Abbott RD, Rogol AD, Evans WS, et al. Relationship between age, percentage body fat, fitness, and 24-hour growth hormone release in healthy young adults: effects of gender. J Clin Endocrinol Metab 1994; 78: 543-8.
- Pasarica M, Zachwieja JJ, Dejonge L, Redman S, Smith SR. Effect of growth hormone on body composition and visceral adiposity in middle-aged men with visceral obesity. J Clin Endocrinol Metab 2007; 92: 4265-70.
- Veldhuis JD, Iranmanesh A, Ho KK, Waters MJ, Johnson ML, Lizarralde G. Dual defects in pulsatile growth hormone secretion and clearance subserve the hyposomatotropism of obesity in man. J Clin Endocrinol Metab 1991; 72: 51-9.
- Rasmussen MH, Hvidberg A, Juul A, Main KM, Gotfredsen A, Skakkebaek NE, et al. Massive weight loss restores 24-hour growth hormone release profiles and serum insulin-like growth factor-I levels in obese subjects. J Clin Endocrinol Metab 1995; 80: 1407-15.
- Mekala KC, Tritos NA. Effects of recombinant human growth hormone therapy in obesity in adults: a meta analysis. J Clin Endocrinol Metab 2009; 94: 130-7.
- Moreno M, Manrique M, Guzmán S, Maiz A, Patiño C, Valdés R, et al. [Change in metabolic risk factors in obese patients in treatment]. Rev Med Chile 2000; 128: 193-200.
- Moulin de Moraes CM, Mancini MC, de Melo ME, Figueiredo DA, Villares SM, Rascovski A, et al. Prevalence of subclinical hypothyroidism in a morbidly obese population and improvement after weight loss induced by Roux-en-Y gastric bypass. Obes Surg 2005; 15: 1287-91.
- Wesche MF, Wiersinga WM, Smits NJ. Lean body mass as a determinant of thyroid size. Clin Endocrinol (Oxf) 1998; 48: 701-6.
- Radetti G, Kleon W, Buzi F, Crivellaro C, Pappalardo L, di Iorgi N, et al. Thyroid function and structure are affected in childhood obesity. J Clin Endocrinol Metab 2008; 93: 4749-54.
- Vella V, Sciacca L, Pandini G, Mineo R, Squatrito S, Vigneri R, et al. The IGF system in thyroid cancer: new concepts. Mol Pathol 2001; 54: 121-4.
- Rezzonico J, Rezzonico M, Pusiol E, Pitoia F, Niepomniszcze H. Introducing the thyroid gland as another victim of the insulin resistance syndrome. Thyroid 2008; 18: 461-4.
- Galofre JC, Pujante P, Abreu C, Santos S, Guillen-Grima F, Fruhbeck G, et al. Relationship between Thyroid-Stimulating Hormone and Insulin in Euthyroid Obese Men. Ann Nutr Metab 2008; 14; 53: 188-94.
- Fazylov R, Soto E, Cohen S, Merola S. Laparoscopic Roux-en-Y gastric bypass surgery on morbidly obese patients
with hypothyroidism. Obes Surg 2008; 18: 644-7. - Isidro ML, Penin MA, Nemina R, Cordido F. Metformin reduces thyrotropin levels in obese, diabetic women with primary hypothyroidism on thyroxine replacement therapy. Endocrine 2007; 32: 79-82.
- Blum M, Dolnikowski G, Seyoum E, Harris SS, Booth SL, Peterson J, et al. Vitamin D(3) in fat tissue. Endocrine 2008; 33: 90-4.
- Bell NH, Epstein S, Greene A, Shary J, Oexmann MJ, Shaw S. Evidence for alteration of the vitamin D-endocrine system in obese subjects. J Clin Invest 1985; 76: 370-3.