Miocardiopatía tirotóxica grave. Consideraciones a propósito de un caso clínico
Valeria Suazo R.1,a, José Manuel Castellón L2. y José Manuel López M1.
Severe thyrotoxic myocardiopathy. Report of one case
1Departamento de Endocrinología. Facultad de
Medicina. Pontificia Universidad Católica de
Chile.
2Departamento de Cardiología. Facultad de
Medicina. Pontificia Universidad Católica de
Chile.
aResidente de Endocrinología. Departamento de
Endocrinología. Facultad de Medicina. Pontificia
Universidad Católica de Chile.
Recibido: 02 Septiembre 2011
Aceptado: 21 Septiembre 2011
We report a previously healthy 43 years old male, that one year ago presented with a hyperthyroidism, treated with metimazole and radioiodine. Two months after receiving the latter, he was admitted to the hospital for dyspnea, tachycardia and chest pain. An atrial fibrillation with a frequency of 190 beats per minute was found. During hospital stay, the patient suffered a cardiogenic shock that recovered. The patient was discharged five days after admission. During follow up, there was a progressive reduction of cardiac symptoms.
Key words: Basedon-graves disease, miocardiopathy, hyperthyroidism.
El compromiso miocárdico en el hipertiroidismo puede
manifestarse como trastornos del ritmo, derrame pericárdico,
insuficiencia cardíaca y/o explicitación de
una cardiopatía preexistente.
Comunicamos el caso de un hombre joven, sin patología
cardíaca conocida, que presentó hipertiroidismo por enfermedad
de Basedow-Graves complicado con grave compromiso
cardíaco evidenciado en el inicio del hipotiroidismo
post tratamiento con I131.
A la luz de esta experiencia clínica, se analizan los componentes
fisiopatológicos, clínicos y terapéuticos relacionados
al compromiso cardíaco en el hipertiroidismo.
Caso clínico
Hombre de 43 años de edad, deportista, sin antecedente de patología cardíaca ni enfermedad reumática, que consulta en diciembre de 2010, por sintomatología de un año de evolución, lentamente progresiva, caracterizada por cansancio fácil, principalmente en extremidades inferiores y temblor fino de manos. La tolerancia térmica era adecuada, sin alteración del sueño, con baja de peso no cuantificada. Su madre fue tratada por enfermedad de Basedow-Graves y un hermano por hipotiroidismo. Reconoce ser fumador de 15 cigarrillos/d.
Al examen físico destacaba peso: 77 kg, pulso 128 x´, regular, presión arterial normal, signo de von Graeffe positivo, ausencia de exoftalmo, piel sudorosa con dermografismo, temblor fino de manos, glándula tiroides crecida, de 60 g, difusa y simétrica; la fuerza de extremidades estaba muy disminuida.
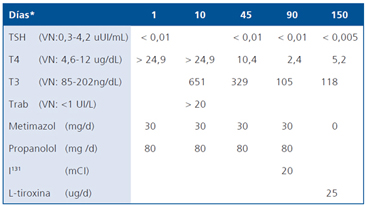
En su primera consulta aportaba los siguientes exámenes: TSH: <0,01 (VN: 0,3-4,2 uUI/mL), T4: >24,9 (VN: 4,6- 12 ug/dL), T3: 631 (VN: 84,6-201,8 ng/dL), Trab >20 (VN:<1 UI/L), Colesterol total 127 (VN <200 mg/dL), GGT 65 (VN 4 - 50 U/L). Con el diagnóstico de enfermedad de Basedow- Graves, se inicia tratamiento con metimazol (30 mg/d) y propanolol (80 mg/d).
La Tabla 1 muestra la evolución inicial del cuadro clínico.
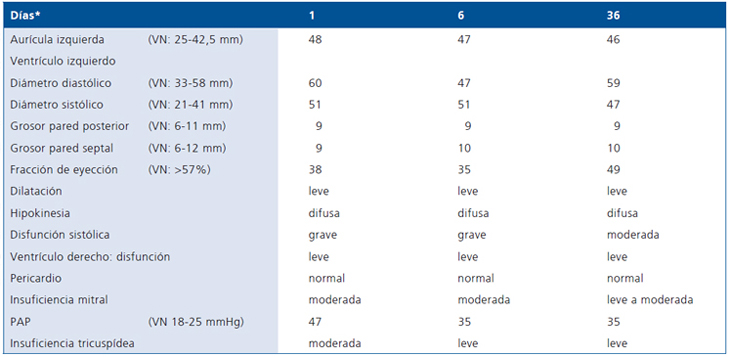
El paciente mejora clínicamente con el tratamiento con metimazol, recupera 9 kg de peso y el tiroides disminuye a 40g. Dos meses después de haber recibido I131, consulta por disnea progresiva de pequeños esfuerzos, ortopnea, palpitaciones rápidas y sensación de opresión retroesternal no dolorosa, intermitente, independiente de los esfuerzos. Se comprueba fibrilación auricular con respuesta ventricular rápida y signos evidentes de insuficiencia cardíaca congestiva, lo que motiva su hospitalización de urgencia. En ese ingreso destacaba buen estado nutritivo y de hidratación, pulso 190 x` en arritmia completa. Presión arterial 120/90 mm/Hg, piel sudorosa con dermografismo, temblor distal de extremidades con disminución de fuerzas. No existía proptosis ocular, pero sí, signo de von Graeffe; el tiroides estaba difusamente aumentado de tamaño. El choque de la punta era hiperdinámico con las yugulares ingurgitadas, taquipnea, con estertores basales en campos pulmonares y edema blando de extremidades inferiores. La radiografía de tórax señalaba cardiomegalia moderada con aorta elongada y congestión pulmonar grave; el ecocardiograma mostraba dilatación y disfunción sistólica grave (Tabla 4).
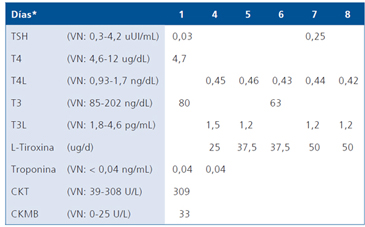
Se inicia tratamiento de urgencia con cedilanid 0,4 mg iv, propanolol 1 mg iv y 40 mg vía oral. Tres horas después presenta edema pulmonar agudo y shock cardiogénico que requiere el uso de BiPAP, dobutamina, milrinona, furosemida, amiodarona, anticoagulación y nitroglicerina endovenosa, observándose una lenta, pero progresiva buena respuesta clínica.
La Tabla 2 resume la evolución de la función tiroidea y los exámenes bioquímicos cardiológicos a partir de la hospitalización de urgencia.
Al cabo de 5 días, estando en mejores condiciones, persiste la fibrilación auricular y es dado de alta manteniendo la anticoagulación oral, e indicación de carvedilol 37,5 mg/d, ramipril 2,5 mg/d, furosemida 40 mg/d, espironolactona 25 mg/d, amiodarona 200 mg/d, eutirox 50 ug/d y magnesio 85 mg/d.
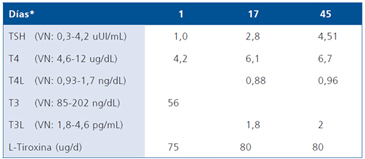
Los controles ambulatorios mostraron mejoría sintomatológica, ausencia de insuficiencia cardíaca, pero con persistencia de la fibrilación auricular con frecuencia ventricular de 70 x’, normotensión y eutiroidismo con el reemplazo ascendente de L-tiroxina. Esta evolución se resume en las Tablas 3 y 4.
Tabla 1. Respuesta de la función tiroidea al tratamiento
*A contar de la primera consulta con uno de los autores.
Tabla 2. Evolución de la función tiroidea y marcadores bioquímicos cardiológicos
*A contar del ingreso a la unidad de paciente crítico.
Tabla 3. Función tiroidea en los controles ambulatorios post hospitalización
*A contar de la fecha de alta desde la unidad de paciente crítico.
Tabla 4. Evolución ecocardiográfica
*A contar desde el ingreso a la unidad de paciente crítico.
Discusión
El sistema cardiovascular puede ser afectado por diferentes trastornos endocrinológicos, especialmente alteraciones de la función tiroidea, paratiroidea, suprarrenal y tumores secretantes como carcinoides, feocromocitoma y acromegalia1,2.
Las hormonas tiroideas participan en la regulación de la frecuencia cardíaca, el tono y la contractibilidad vascular y miocárdica y regulan las demandas metabólicas del organismo. Las manifestaciones cardiovasculares de las enfermedades tiroideas habitualmente son reversibles al recibir tratamiento oportuno y eficaz1.
En el caso del hipertiroidismo, el exceso de hormonas
tiroideas genera un estado cardiovascular hiperdinámico, cuyas
manifestaciones relevantes son:
a) Disminución de la resistencia vascular sistémica.
b) Taquicardia.
c) Aumento del gasto cardíaco (mayor volumen de eyección
y de la contractibilidad miocárdica).
d) Aumento del consumo de oxígeno miocárdico y mayor
demanda coronaria1-3.
a) Disminución de la resistencia vascular sistémica: la vasodilatación periférica ocurre en razón de la rápida utilización de oxígeno con aumento de productos finales del metabolismo y relajación del musculo liso vascular. La vasodilatación explica en un 50 a 60% la disminución de la resistencia vascular sistémica. Este hecho es central en relación a los otros cambios del hipertiroidismo: taquicardia, aumento de perfusión a la piel, músculos esqueléticos y corazón y aumento de la presión arterial diferencial que se manifiesta en un pulso céler. El hecho que el flujo sanguíneo renal no aumente concomitantemente con la vasodilatación sistémica activa el eje renina-angiotensina-aldosterona que genera aumento del volumen intravascular1-3.
b) Taquicardia: las palpitaciones están presentes en el 85% de los hipertiroideos. Las arritmias cardíacas son comunes, especialmente los extrasístoles auriculares y ventriculares y la fibrilación auricular. Esta última, en personas mayores puede ser la única manifestación clínica de tirotoxicosis (hipertiroidismo apático). Aproximadamente 10 a 20% de los pacientes con fibrilación auricular tienen tirotoxicosis y hasta un 20% de los con tirotoxicosis presentan fibrilación auricular. El riesgo de tromboembolismo arterial está aumentado. Otras arritmias auriculares son inusuales. En general, las arritmias ventriculares trasuntan una enfermedad cardíaca de base1-3.
c) Aumento del gasto cardíaco: la contracción sistólica y la relajación diastólica están aumentadas en el hipertiroidismo, haciendo que el corazón funcione cercano al nivel de su máxima capacidad, con disminución evidente de su reserva1.
En el hipertiroidismo, los niveles de catecolaminas están normales o bajos; la respuesta hiperadrenérgica se explicaría por aumento del número y sensibilidad de los receptores simpáticos β inducido por las hormonas tiroideas. La intolerancia al ejercicio y la disnea de esfuerzos pueden ocurrir con o sin insuficiencia ventricular izquierda. La insuficiencia cardíaca generalmente es precipitada por fibrilación auricular que perjudica el llenado diastólico. Muchos de los pacientes con insuficiencia cardíaca pueden tener una cardiopatía de base que los predispone a desarrollar disfunción ventricular4. La incidencia de prolapso de la válvula mitral está aumentada en los pacientes con enfermedad de Basedow-Graves, lo que se explicaría por los cambios hemodinámicos descritos y además por una posible predisposición genética1,4. Nuestro paciente presentó fibrilación auricular e insuficiencia cardíaca global con muy mala respuesta a los betabloqueadores a pesar de la evidente hiperdinamia, llegando a desarrollar shock cardiogénico.d) Aumento del consumo de oxígeno miocárdico y mayor demanda coronaria: frecuentemente la tirotoxicosis es un factor precipitante de angina en pacientes con cardiopatía coronaria conocida o no. La angina mejora cuando se trata la tirotoxicosis y el infarto miocárdico es raro5.
Acciones a nivel molecular de las hormonas tiroideas
Los cambios cardiovasculares que ocurren en el hipertiroidismo resultan de la acción molecular de las hormonas tiroideas en el corazón y vasos sanguíneos. El efecto cardíaco es mediado por un mecanismo dual, genómico y no genómico.Por su naturaleza lipofílica, T3 y T4 difunden fácilmente al interior de las células diana, incluidos los cardiomiocitos. Aunque es conocido que T4 se convierte en T3 en el citosol de la mayoría de las células, no hay evidencia de que esto ocurra también en los cardiomiocitos. T3 se une a los receptores nucleares específicos, unión que activa al receptor nuclear, el cual se homodimeriza o heterodimeriza con el receptor del ácido retinoico (RXR). Tanto el complejo de T3 con dos receptores específicos o la forma activada T3/receptor específico/RXR reconocen algunas de las secuencias de ADN, conocidas como “elementos de respuesta tiroidea” (ERT), ubicadas en la región promotora de genes blanco. Después de la unión del complejo señalado con un ERT, el promotor del gen objetivo se activa lo que deriva en el inicio de la transcripción. Los genes cardíacos reconocidos como objeto de activación transcripcional son: la cadena pesada α de la miosina (MHC-α), las enzimas calcio-ATPasa del retículo sarcoplásmico (SERCA) y Na-K-ATPasa, el receptor adrenérgico β1, la troponina I y el péptido natriurético auricular. A la inversa, la transcripción de otros genes es reprimida, como es el caso de la cadena pesada β de miosina MHC-β y el fosfolamban. El resultado final sobre la contractilidad miocárdica representa el balance diferencial en la expresión de los genes de las cadenas pesadas de miosina. En el músculo ventricular se han identificado tres isoformas de miosina: V1 (cadenas pesadas α-α, MHC αα), V2 (cadenas pesadas α y β, MHC αβ) y V3 (cadenas pesadas ββ, MHC ββ). La hormona tiroidea, a través de su actividad transcripcional en MHC-α y MHC-β cambia la expresión de la isoformas mediante el aumento de la síntesis de V1 y disminución de V3. La isoforma V1 tiene mayor actividad enzimática ATPasa y acelera el acortamiento de la fibra muscular3-6.
Otro posible mecanismo genómico dice relación con la liberación de calcio y su recaptación por el retículo sarcoplásmico, lo que regula la frecuencia de acortamiento y relajación de la fibra miocárdica. A través de la transcripción genómica positiva de SERCA y de la disminución de la proteína fosfolambam, las hormonas tiroideas aumentan la recaptación de calcio por el retículo sarcoplásmico, lo que, a su vez, aumenta el acmé de tensión de los cardiomiocitos y disminuye el tiempo de contracción muscular ventricular. Este mecanismo también explica la mayor capacidad de relajación diastólica propia del corazón en situación de hipertiroidismo. Se ha sugerido que el aumento del inotropismo podría resultar del mayor número y sensibilidad de los receptores β adrenérgicos cardíacos. Datos más recientes, han demostrado que las hormonas tiroideas no aumentan la sensibilidad del ventrículo izquierdo a la estimulación betaadrenérgica3- 6.
Las vías no genómicas, contrariamente a las genómicas, son de respuesta rápida y explican acciones tales como el aumento del gasto cardíaco después de la inyección intravenosa de T3. A través de vías no genómicas, las hormonas tiroideas prolongan la inactivación de los canales de sodio en los cardiomiocitos, aumentan el consumo de sodio intracelular y el intercambio entre sodio y calcio en el retículo sarcoplásmico, lo que puede explicar la actividad inotrópica aguda de T33-6.
La Figura 1 muestra de manera esquemática, el mecanismo
genómico de acción de las hormonas tiroideas en el
cardiomiocito.
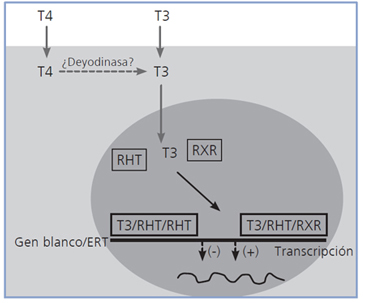 Figura 1. Mecanismo genómico de acción de las hormonas tiroideas
en el cardiomiocito. RHT: receptor de hormona tiroidea. RXR
receptor de ácido retinoico. ERT: elemento de respuesta tiroidea.
Figura 1. Mecanismo genómico de acción de las hormonas tiroideas
en el cardiomiocito. RHT: receptor de hormona tiroidea. RXR
receptor de ácido retinoico. ERT: elemento de respuesta tiroidea.
El tratamiento del hipertiroidismo está dirigido a mejorar rápidamente los síntomas y reducir la demanda sobre el corazón; se basa en el bloqueo de la formación y liberación de hormonas tiroideas con drogas antitiroideas, seguido de la ablación glandular con I131. Los bloqueadores-β son ampliamente usados como tratamiento sintomático2. En pacientes con insuficiencia cardíaca, si la taquicardia se transforma en un efecto deletéreo y no compensador, puede usarse esmolol endovenoso, de rápido comienzo de acción y corta vida media7. El propanolol agrega un buen efecto bloqueador de la conversión periférica de T4 a T3. La dosis puede ser titulada de acuerdo a la frecuencia cardíaca; usualmente se usan 20 a 40 mg hasta 4 veces al día2.
Las tionamidas bloquean la síntesis y la liberación de hormonas tiroideas a través del bloqueo de la oxidación, organificación y acoplamiento del yodo a la tirosina. El metimazol ofrece como ventaja la comodidad de administración (1 vez al día) y la seguridad (menos incidencia de efectos adversos comparado con el propiltiouracilo). Depleta los depósitos intratiroideos de hormonas y evita una eventual crisis tirotóxica en relación a la tiroiditis actínica post ablación con I131. La dosis habitualmente usada fluctúa entre 10-30 mg/d2.
Otras drogas como el yodo, que bloquea la liberación de hormona tiroidea y debe ser usada concomitantemente con tionamidas, y los glucocorticoides, que disminuyen la liberación de hormonas tiroideas y la conversión periférica de T4 en T3, se reservan para condiciones de extrema gravedad como la tormenta tiroidea, insuficiencia cardíaca grave o angina inestable secundaria a tirotoxicosis2.
El I131 es el tratamiento definitivo de elección para los pacientes con enfermedad de Graves, el cual resuelve el hipertiroidismo en un plazo de 3 a 6 meses; algunos pacientes requieren tionamidas durante este período y la mayoría, sustitución tiroidea por hipotiroidismo2.
El tratamiento de la insuficiencia cardíaca congestiva y la fibrilación auricular no difiere del propio de pacientes eutiroideos. Incluye bloqueo adrenérgico beta no selectivo (propanolol), o selectivo para controlar la taquicardia8. Frecuentemente los pacientes son relativamente resistentes a la digoxina7. El tratamiento de la fibrilación auricular debe limitarse inicialmente al control de la frecuencia cardíaca, ya que la cardioversión no será exitosa o perdurable si no se corrige la tirotoxicosis. En el curso de las 6 a 8 semanas de alcanzado el eutiroidismo, no es raro que ocurra espontáneamente la cardioversión a ritmo sinusal (60%), situación más improbable de ocurrir en pacientes de edad avanzada. La anticoagulación debe ser mantenida hasta que se obtenga el ritmo sinusal y el eutiroidismo8.
En nuestro paciente el tratamiento del hipertiroidismo fue convencional con metimazol (30 mg/d) y propanolol (40 mg c/12 h) con buena respuesta, en los plazos habituales. Después de 14 semanas, recibió 20 mCI de I131, dosis justificada por el tamaño tiroideo, el nivel de hormonas tiroideas y lo sintomático del cuadro. A partir de las 8 semanas de recibir I131, se evidencia hipotiroidismo con TSH aún suprimida (Tabla 2); es en este período cuando se manifiesta la sintomatología y la signología de insuficiencia cardíaca global que requiere hospitalización de urgencia. El largo tiempo de tirotoxicosis grave sin tratamiento consumió, probablemente, la reserva cardíaca de un individuo joven, supuestamente sano, y ello se presentó paradojalmente en el inicio del período de hipotiroidismo post tratamiento con I131. La mantención de signos de dilatación ventricular en el ecodoppler cardíaco, 8 semanas después de alcanzado el eutiroidismo, pone una nota de cautela respecto de la condición de normalidad cardiológica total basal del paciente y de su total recuperabilidad cardiovascular. Sólo el control a largo plazo ayudará a una mejor clarificación del problema.
Referencias
- Vela B. S, Crawford Michael H, Current Diagnosis & Treatment:
Cardiology, 3e: “Chapter 32. Endocrinology & the Heart”.
Disponible en: http://www.accessmedicine.com/content.
aspx?aID=3650648 (consultado el 22 de Julio de 2011).
- Mandel S, Larsen P, Davies T. 2011. Thyrotoxicosis. En: Williams
Textbook of Endocrinology (12th Ed.). Philadelphia. Elsevier,
p. 362-399.
- Bahaa M. Fadel, Samer Ellahham. 2000. Hyperthyroid heart
disease. Clinical Cardiology 23: 402-408.
- Klein I, Ojamaa K. 2001. Thyroid hormone and the cardiovascular
system. New England of Medicine 344: 501-509.
- Klein I., Danzi S. 2007. Thyroid disease an the heart. Circulation
116: 1725-1735.
- Arsanjani R, McCarren M, Bahl J. 2011. Goldman S., Translational
potencial of thyroid hormone and its analogs. Journal of Molecular
and Celular Cardiology. Article in press.
- Su-yin Ngo, A. 2006. Thyrotoxic heart disease. Resuscitation 70:
287-290.
- Shimizu T, Koide S, Yoshimura J, Kiminori S, Ito K, Nakasawa H.
2002. Hyperthyroidism and the Management of Atrial Fibrillation.
Thyroid 12: 489-493.
- Bird-Lake E. 2011. Severe transient left ventricular dysfunction
induced by thyrotoxicosis. Netherland Heart Journal 19: 352-354.
- Ching Soh M., Croxson M. 2000. Fatal thyrotoxic cardiomyophaty
in a young man. BMJ 338: 476-477.
- Froeschl M, Haddad H. 2005. Thyrotoxicosis an uncommon cause of heart failure. Cardiovascular Pathology 14: 24-27.