Rev. chil. endocrinol. diabetes 2019; 12 (1) Volver a Índice
Libro de Resúmenes Congreso 2018
Congreso Chileno de Endocrinología y Diabetes
8-10 de noviembre de 2018
Coquimbo, Chile
Casos Clínicos y Orales
C 1. PARATIROIDECTOMÍA TRANSORAL ENDOSCÓPICA POR ACCESO VESTIBULAR ORAL. CIRUGÍA SIN CICATRIZ. PRIMERA SERIE EN SUDAMÉRICA
Patricio Cabané Toledo1, Patricio Gac Espinoza2, Francisco Rodríguez Moreno2, Daniel Rappoport Wurgaft2
1. Hospital Clínico Universidad de Chile, Clínica Indisa. 2. Hospital Clínico Universidad de Chile. Santiago, Chile.
Contenido: El abordaje quirúrgico de elección de tiroides y paratiroides, ha sido clasicamente la cervicotomía transversa. En el hiperparatiroidismo primario (HPT1rio), con la optimización de los métodos de localización preoperatoria, se han creado técnicas abiertas mínimamente invasivas con mejor resultado estético y menor tiempo de recuperación (videoasistida, endoscópica transaxilar, retroauricular, etc.). Con el objetivo de eliminar las cicatrices externas se ha implementado una nueva técnica quirúrgica que utiliza abordajes endoscópicos a través de orificios naturales (NOTES). En los últimos años se ha hecho conocida la técnica de tiroidectomía y paratiroidectomíatransoral endoscópica por el vestíbulo oral (TOETVA y TOEPVA).
Técnica: Anestesia general (intubación nasotraqueal), incisiones (3) en vestibulo oral, hidrodisección, dilatación subcutánea hasta hueco
supraesternal, introducción de trocares (uno de 10 mm y dos de 5 mm). Insuflación con CO2 hasta 6 mmHg. Disección subplatismal, Acceso
por rafe, retracción de músculos pretiroideos con punto transcutáneo. Disección extracapsular de tiroides electro bisturí y energías avanzadas.
Identificación de nervio recurrente y adenoma. Resección y extracción por trocar de 10 mm.
Hemostasia y Cierre
Caso 1. Paciente de 65 años con HPT 1rio con nódulo hiperfuncionante superior derecho (calcemia 10,8 mg/dl, PTH: 230 pg/ml, Fosfemia: 2,1
mg/dl) Se realiza paratiroidectomíatransoral endoscópica por abordaje vestibular. Resección adenoma superior derecho, PTH intraop: Basal:
180 pg/ml; 15 min: 42,6 pg/ml; 30 min: 33,4 pg/ml. Tiempo quirúrgico: 180 min. Sin hipocalcemia ni disfonía. Alta a las 48 hrs.
Caso 2. Pacte 33 años, HPT 1rio con PTH preop: 520 pg/ml y calcemia 11 mg/dl. Ubicación inferior derecha por ultrasonografía y sestamibi.
Resección transoral sin complicaciones. Adenoma de 2 cm. PTH intraop: Basal: 549 pg/ml; 15 min: 31 pg/ml; 30 min: 21,9 pg/ml. Tiempo
quirúrgico 120 min. Sin hipocalcemia ni disfonía PO. Alta a las 24 hrs.
La técnica TOEPVA se realiza sin incidentes con buena evolución postoperatoria. Ha demostrado ser una técnica segura, con resultados
similares a los de la técnica abierta, pero sin dejar cicatrices visibles.
Financiamiento: Sin financiamiento.
C 2. ABSCESO DE QUISTE DE LA BOLSA DE RATHKE: PRESENTACIÓN DE CASO CLÍNICO
Francisco Muñoz Ortíz2, Flavia Nilo Concha1, Francisco Guarda Vega2, Pablo Villanueva Garín2, Claudio Callejas Cánepa2
1. Hospital Clínico Universidad Católica. 2. Pontificia Universidad Católica de Chile. Santiago, Chile.
Contenido: El absceso hipofisario es una patología rara que puede presentarse en una glándula sana, con lesiones selares preexistentes
(adenoma, quiste de la bolsa de Rathke, craneofaringioma) o con intervenciones previas (cirugía, radioterapia). Típicamente se presenta con
cefalea, hipopituitarismo, diabetes insípida y hallazgos característicos en la resonancia magnética (RM), habitualmente en ausencia de fiebre
e inflamación sistémica.
Se presenta el caso de una paciente con el antecedente de un quiste de la bolsa de Rathke operado, que evoluciona con un absceso hipofisario.
Caso clínico: Mujer de 51 años con antecedente de quiste de la bolsa de Rathke operado en el año 2004, sin remanente visible y eupituitárica
hasta el año 2013. El 2016 consultó por cefalea, alteración visual y baja de peso, con RM que mostró lesión quística de 16 mm que contactaba
vía óptica sin alteración campimétrica, por lo que se mantuvo en observación, con resolución espontánea en RM de abril 2018.
En junio 2018 consulta por cefalea y diplopía por paresia de III nervio craneano derecho de pocos días de evolución, afebril, sin otros hallazgos clínicos. Se realiza RM que mostró 2 lesiones quísticas hipointensas en T1, hiperintensas en T2, con una cápsula gruesa que se refuerza con gadolinio y con contacto de vía óptica; además se objetiva hipopituitarismo sin diabetes insípida, campo visual poco interpretable por poca cooperación en contexto de una infección respiratoria intercurrente y PCR levemente alta.
Ante la sospecha de un absceso hipofisario se hospitaliza y previa preparación con hidrocortisona y levotiroxina, se realiza cirugía transesfenoidal endoscópica donde se objetiva un absceso con cápsula indurada y salida de material purulento del esfenoides y ambas cavidades quísticas, además de un defecto en el piso selar. Los cultivos confirmaron infección por Staphilococcusaureus meticilino sensible, por lo que recibió cloxacilina durante 3 semanas. La paciente evoluciona con diabetes insípida post-operatoria, remisión de sus síntomas, PCR normal y RM que mostró ausencia de lesiones residuales. Queda pendiente la reevaluación del hipopituitarismo en diferido.
Conclusión: El absceso hipofisario es una causa rara dentro de las lesiones selares quísticas, que puede aparecer en glándulas sanas o
con alteraciones estructurales previas como este caso. Habitualmente debuta con cefalea, hipopituitarismo y diabetes insípida en ausencia
de fiebre, signos de irritación meníngea o elevación de parámetros inflamatorios. Si bien clínicamente puede confundirse con otras patologías
selares, sus hallazgos en RM son característicos y ayudan al diagnóstico diferencial. El tratamiento habitual comprende el drenaje quirúrgico
por vía transesfenoidal y el uso de antibioterapia endovenosa prolongada, además del manejo del hipopituitarismo frecuentemente asociado.
Financiamiento: Sin financiamiento
C 3. HIPERCALCEMIA ASOCIADA A MIOSITIS GRANULOMATOSA SARCOIDEA SÍMIL, UN CUADRO INFRECUENTE A CONSIDERAR
Thomas Uslar Nawrath1, Gilberto González Vicente2, Roberto Olmos Borzone2, Jaime Godoy Santin3, Patricio Mellado Talesnik3
1. Departamento de Medicina Interna, Hospital Clínico Pontificia Universidad Católica de Chile. 2. Departamento de Endocrinología, Hospital Clínico Pontificia
Universidad Católica de Chile, 3Departamento de Neurología, Hospital Clínico Pontificia Universidad Católica de Chile. Santiago, Chile.
Contenido: La hipercalcemia asociada a enfermedades granulomatosas está bien descrita y su mecanismo subyace en la expresión extrarrenal
de 1-alfa-hidroxilasa en macrófagos activados con producción no regulada de 1,25OHD independiente de PTH. La sarcoidosis es una de las
enfermedades más representativas para este mecanismo de hipercalcemia, pero existen múltiples otras. Presentamos un caso de hipercalcemia
asociada a enfermedad granulomatosa para ilustrar una entidad recientemente descrita.
Caso clínico: Paciente de 87 años consultó por 3 meses de tetraparesia de predominio proximal asociado a baja de 10k y astenia luego de viaje
al Caribe. Al examen físico: tetraparesia proximal simétrica con reflejos conservados, sin otros hallazgos. Al laboratorio destacó calcemia de
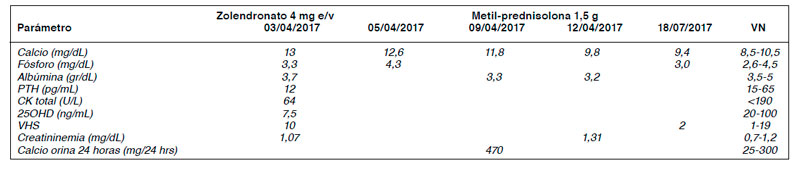
13,4 mg/dL, PTH 12 pg/mL y CK normal (Tabla 1). PET/CT-FDG evidenció aumento de metabolismo glucídico intenso y difuso en musculatura
proximal, sin linfadenopatíasmediastínicas u otros hallazgos. Se realizó biopsia deltoidea que describió granulomasepiteloideos no caseificantes
ni necrotizantes, compatible con enfermedad granulomatosa muscular. Cuantiferon TBC negativo y estudio de causas neoplásicas negativo. Se diagnosticó miositis granulomatosa sarcoidea símil. Se manejó con Zolendronato 4 mg e/v y dosis alta de Prednisona con normalización de
calcemia y tetraparesia. Al 3er mes, con dosis decreciente de Prednisona, persiste con normocalcemia y sin paresia.
Comentario: La miositis granulomatosa sarcoidea símil es una entidad rara (< 10 casos publicados) y recientemente descrita como una reacción sarcoidea muscular en ausencia de cuadro multisistémico concordante con sarcoidosis. A diferencia del compromiso muscular en sarcoidosis, este se presenta con debilidad muscular sin mialgia, suele cursar con CK total normal, captación muscular difusa en PET/CT-FDG, e hipercalcemia moderada a grave independiente de PTH, con 1,25OHD elevado. Nuestro paciente inició sus síntomas luego de fuerte exposición solar, lo cual probablemente descompensó su cuadro y es compatible con la fisiopatología involucrada. Los hallazgos histopatológicos de las miositis granulomatosas son inespecíficos, por lo que el diagnóstico diferencial debe ser amplio. Creemos que esta nueva entidad debe ser incorporada dentro del diagnóstico diferencial de hipercalcemia PTH independiente, con especial énfasis en el uso de PET/CT-FDG para una correcta aproximación.
C 4. METÁSTASIS HIPOFISARIA ASOCIADA A ACROMEGALIA ECTÓPICA POR CARCINOMA PULMONAR DE CÉLULAS PEQUEÑAS: IMPORTANCIA DEL ANÁLISIS CLÍNICO-IMAGENOLÓGICO EN LESIONES SELARES
Raiza García Lois1, Pablo Villanueva Garín2, Flavia Nilo Concha3, Héctor Galindo Aranibar4, Sebastián Mancilla Wistuba5, Francisco Guarda Vega3
1. Departamento Endocrinología, Pontificia Universidad Católica de Chile. 2.
Departamento de Neurocirugía, Programa de Tumores Hipofisarios UC, Pontificia Universidad Católica de Chile. 3. Departamento de Endocrinología, Programa de Tumores Hipofisarios UC, Pontificia Universidad Católica de Chile. 4. Departamento de Hematología y Oncología, Pontificia Universidad Católica de Chile. 5. Escuela de Medicina, Pontificia Universidad Católica de Chile. Santiago, Chile.
Introducción: Las características imagenológicas en resonancia magnética (RM) son claves para orientar el estudio etiológico de una lesión hipofisaria. Si bien los adenomas son la causa más frecuente, rasgos atípicos como la posición de la hipófisis normal, anatomía del diafragma selar y presencia de compromiso de tallo y neurohipófisis permiten extender el diagnóstico diferencial y ampliar el estudio hormonal y etiológico. A raíz de un caso clínico, presentamos la importancia de conocer las características imagenológicas que deben hacer sospechar diagnósticos alternativos.
Caso clínico: Mujer de 57 años con antecedentes de HTA, hipotiroidismo primario y tabaquismo, consultó por alteración de campo visual de 1 mes de evolución. Se evidenció un defecto bitemporal en la campimetría, por lo que se realiza RM selar que muestra una lesión en reloj de arena isointensa en T1, hipointensa en T2, con captación heterogénea de gadolinio, compromiso de pars tuberalis y tallo de 18mm, compresión quiasmática e hipófisis normal desplazada hacia inferior y lateral; informado como macroadenoma hipofisario. Al examen destacaba facies tosca sin otros estigmas de acromegalia e historia de poliuria y nicturia significativas. Estudio hormonal confirmó acromegalia, hiperprolactinemia leve y una probable diabetes insípida (Prolactina 74.8ng/mL, IGF-1 215ng/mL (VN 36- 200), nadir GH 1.87ng/mL, FSH 12.7mUI/mL, TSH 3.41uUI/mL, T4 7.6ng/dL, Cortisol basal 9.1 y post ACTH 19ug/dL, Na 143mEq/L, Osm plasmática 316mOsm/ Kg, Osm urinaria 493mOsm/Kg).
Debido a imagen atípica para adenoma por el desplazamiento inferior de hipófisis normal, compromiso de tallo y preservación de diafragma selar asociado a posible diabetes insípida, se sospechó lesión no adenomatosa. Se realizó TC de tórax, abdomen y pelvis que evidenció 3 nódulos pulmonares mal delimitados asociado a múltiples adenopatías hiliares, subcarinales y precarinales, con metástasis hepática y suprarrenal izquierda. Biopsia transbronquial confirmó carcinoma pulmonar de células pequeñas con probables metástasis hipofisaria y acromegalia por producción ectópica de GHRH. Se trató con quimioterapia con Etopósido + Cisplatino + Pembrolizumab/placebo y Radioterapia paliativa holocránea 30 Gy en 10 fracciones. Presentó buena respuesta estructural de lesión hipofisaria y de enfermedad sistémica, sin embargo al 5° mes de tratamiento se pesquisó progresión cerebral con múltiples nuevas lesiones nodulares supra e infratentoriales.
Conclusión: Este caso nos recuerda la importancia de la evaluación crítica de la clínica y RM frente a una lesión selar, considerando elementos como las
características radiológicas, la posición de la hipófisis normal, preservación del diafragma selar y presencia de compromiso de tallo y de diabetes insípida. La
sospecha clínica en lesiones atípicas puede cambiar radicalmente el diagnóstico y manejo como este caso.
Financiamiento: No.
C 5. ADRENOLEUCODISTROFIA LIGADA AL X: PRESENTACIÓN DE TRES CASOS
Susana González Catalán1, María Ochoa Molina1, Alejandro Martínez Aguayo1, Claudia Godoy Cortés1
1. Pontificia Universidad Católica de Chile. Santiago, Chile.
Contenido: La adrenoleucodistrofia ligada a X (X-ALD) es una enfermedad neurodegenerativa progresiva, por una alteración en el gen ABCD1 ubicado en el cromosoma Xq28 que produce la deficiencia de ALDP por lo que se altera la β-oxidación peroxisomal y produce una acumulación de los ácidos grasos de cadena muy larga (AGCML) en suero, la corteza adrenal y sustancia blanca del sistema nervioso central. La incidencia 1 en 17,000 recién nacidos vivos.
Caso 1: Varón de 7 años 10 meses con estrabismo se realiza RM cerebral: muestra signos de leucodistrofia. Asintomático. Estudio de AGCML: elevados. Padres y hermana sanos. ACTH 387 pg/ml (1.1-3.8), Actividad de renina 3.10 ng/mL/Hr (1.1-3.8), cortisol basal 12.7 ug/dl (3-15.4) y post ACTH 14.1 ug/dL. Se inicia hidrocortisona, se mantiene estable por 2 años, luego compromiso neurológico progresivo: no sujeta cabeza, espasticidad y ceguera cortical a los 16 años.
Caso 2: Varón 2 años 3meses, sano, primer embarazo, padres sanos con antecedente familiar de dos tíos maternos con drenoleucodistrofia.
Se realiza estudio que confirma diagnóstico de Adrenoleucodistrofiapresintomática, confirmada por AGCML y mutación c.1175T7C(pleu3P2Pro)
en el gen ABCD1. Actividad de renina 2.41 ng/mL/Hr (1.0-6.5), cortisol 17.6 ug/dl (3-15.4) y post ACTH 30 minutos 22.2 ug/dL. Actualmente, 5
años 3 meses asintomáticos.
Caso 3: Varón 9 años 10 meses, presenta hemiparesia derecha progresiva, se realiza RM cerebral con lesiones de leucodistrofia. Medición de AGCML que son positivos. Paciente evoluciona con deterioro neurológico progresivo y falla adrenal.
La X-ALD es una enfermedad progresiva que tiene una fase asintomática, que evoluciona con compromiso neurológico e insuficiencia
suprarrenal. En la actualidad no existe un tratamiento curativo disponible y el trasplante de células madre hematopoyéticas se ha estudiado
que logra una estabilización de la enfermedad. Es importante buscar la insuficiencia adrenal y la consejería genética.
Financiamiento: Sin financiamiento.
C 6. TRATAMIENTO ADYUVANTE CON INHIBIDOR DE AROMATASA EN EL MANEJO DE RECIDIVA DE TUMOR DE CÉLULAS DE LA GRANULOSA: REPORTE DE UN CASO
Liuba León Hernández1, Raiza García Lois1, Lorena Mosso Gómez1, Paulina Villaseca Délano1, Eugenio Arteaga Urzúa2, Alejandra Martínez García1.
1. Pontificia Universidad Católica de Chile. 2. Hospital Clínico Pontificia Universidad Católica de Chile. Santiago, Chile.
Contenido: Los tumores ováricos de las células de la granulosa (TOCG) son tumores infrecuentes que se caracterizan por su elevada recurrencia. QMT y RDT, tienen eficacia limitada con efectos adversos significativos; y los inhibidores de aromatasa (IA) han surgido como alternativa. Presentamos un caso de recidiva de TOCG productor de Inhibina, tratado con IA con buena respuesta. Caso: Paciente de 46 años, que al año de control por probable amenorrea hipotalámica funcional, se evidencia en ecografía transvaginal, una imagen anexial izquierda sólida de 3 cm, vascularizada al doppler (CEA y CA-125 negativos). TAC abdomen y pelvis, sin otros hallazgos. Fue tratada con histerectomía y salpingooforectomía bilateral. La biopsia diagnostica: TOCG de tipo adulto de 33x23x23 mm con estudio inmunohistoquímico positivo para Inhibina y CD99, y negativo para AE1/AE3 y EMA. Posterior a cirugía, la medición de Inhibina B (IB) fue positiva, con reducción inicial e incremento gradual hasta 102 pg/mL a los 2 años (Tabla 1). Por ello, se realizó RM pelvis que constató adenopatía única ilíaca externa izquierda de 15x10 mm. La cirugía exploratoria demostró implantes a nivel del ligamento infundíbulo-pélvico izquierdo. La biopsia diagnosticó recurrencia de TOCG en el tejido conectivo con inmunohistoquímica positiva para receptores de estrógenos (RE) y progesterona. A las 6 semanas de la cirugía, la IB se mantuvo detectable (Tabla 1). En este contexto, se decidió realizar tratamiento con RDT de pelvis (45Gy en 25 fracciones) y Letrozol (2,5 mg/día). Desde el inicio del IA, hace un año 7 meses, no se ha demostrado recurrencia y las concentraciones de IB se hicieron indetectables. Comentario: No existe un tratamiento estándar para la recurrencia en TOCG. Aunque la evidencia en el uso de IA es variable, se han descrito casos con respuesta transitoria y también persistente.
Más del 90% de las pacientes con TOCG de tipo adulto, tienen una mutación específica del factor de transcripción FOXL2 (405C>G) C134W. FOXL2 mutado, se une al promotor de aromatasa produciendo un “up-regulation” de su actividad, lo cual podría promover el desarrollo y progresión de los TOCG. Por tanto, los IA podrían ofrecer ventajas potenciales en estos casos. Nuestra paciente mantiene una excelente respuesta después de 19 meses de inicio de IA, que es reportada como superior en las pacientes con RE positivos en la inmunohistoquímica, pero su búsqueda no es evaluada de rutina en TOCG. Proponemos el uso de IA como parte del manejo de la recurrencia en TOCG de tipo adulto, y sugerimos incorporar la búsqueda dirigida de RE, ya que su presencia podría favorecer la eficacia del IA a largo plazo.
Financiamiento: Sin financiamiento.
Tabla 1. Niveles plasmáticos de Inhibina-B, FSH y Estradiol.

C 7. PARAGANGLIOMA VESICAL METASTÁSICO CON MUTACIÓN SUCCINATO DESHIDROGENASA B
Carolina Orellana Bravo1, Jesús Véliz López1, René Díaz Torres1, Nelson Wohllk González2. Santiago, Chile.
1. Hospital del Salvador. 2. Hospital del Salvador, Laboratorio IEMA. Santiago, Chile.
Introducción: Los paragangliomas (PGG) son tumores extraadrenales productores de catecolaminas, frecuentemente asociado a síndromes genéticos. Su localización vesical representa el 10% de todos los PGG. Los síntomas de presentación son hematuria macroscópica, cefalea, palpitaciones, hipertensión arterial (HTA) e HTA durante la micción. Su diagnóstico la mayoría de las veces es un desafío y el manejo es muy similar al feocromocitoma. Se presenta el caso de PGG vesical metastásico con mutación succinato deshidrogenasa B (SDHB).
Caso clínico: hombre de 40 años, con antecedentes de HTA desde 2012. Consulta en el 2013 por cuadro de hematuria macroscópica a urología. Se realiza
resección transuretral (RTU) con resección de pólipo vesical y biopsia compatible con PGG. PET/CT 68 Ga-DOTATATE: engrosamiento parietal
derecho de vejiga con sobreexpresión de receptores de somatostatina (SUV máx 30). Sin otras lesiones. Se realiza el 2014 cistectomía parcial
laparoscópica con linfadenectomía pelviana y neoimplante ureteral derecho. Biopsia: PGG vesical que infiltra focalmente grasa perivesical, con
márgenes y adenopatías negativos. Inmunohistoquímica inmunorreactiva para CD56, sinaptofisina y cromogranina A. Evoluciona con remisión
de síntomas. Reinicia controles el 2017 por HTA asociado a palpitaciones y aumento de volumen costal izquierdo. PET/CT 68 Ga-DOTATATE:
recidiva con numerosas lesiones osteolíticas en calota, maxilar, columna vertebral, parrilla costal bilateral, esternón, extremo medial clavícula
izquierda, escápula y húmero derecho, pelvis y en extremos proximales de ámbos fémures compatible con metástasis de tumor neuroendocrino.
Estudio genético: mutación heterocigota SDHB, variante patogénica c.591delC (p.Ser198Alafs*22). Pendiente estudio en familiares. Se indica
terapia con 177Lutecio- DOTATATE, cada ciclo 150 mCi . Exámenes, III ciclo: metanefrinas 125 ug/gr creatinina (VN 25 – 155), normetanefrinas
2657 ug/gr creatinina (VN 46 – 256). Exámenes V ciclo177Lutecio-DOTATATE: metanefrinas 35.7 ug/gr creatinina, normetanefrinas 1478 ug/
gr creatinina. A las 24 horas del quinto ciclo se registran imágenes en sitios similares a las observadas pero de menor intensidad sugiriendo
disminución de la actividad tumoral en relación a la observada en los tratamientos previos. Sin nuevas lesiones. Dosis acumulada (793 mCi).
Paciente evoluciona con control de HTA, sin necesidad de tratamiento antihipertensivo, palpitaciones ocasioanles y dolores óseos. Pendiente
último sesión de 177-Lutecio.
Discusión: El PGG metastásico es una patología muy infrecuente, que plantea un desafío en el diagnóstico
y tratamiento. El estudio genético de los PGG siempre debe ser realizado cuando se hace el diagnóstico y frente a un PGG metastásico la
mutación SDHB es la más probable.
Financiamiento: Sin financiamiento.
C 8. TRASTORNO DE LA DIFERENCIACIÓN SEXUAL POR SÍNDROME DE INSENSIBILIDAD ANDROGÉNICA: NUEVA MUTACIÓN. PRESENTACIÓN DE CASO CLÍNICO
Anahí Yizmeyián Maeso1, Vivian Gallardo Tampier1, Carolina Sepúlveda Rubio1, Soledad Villanueva Toral1, Ana Rocha Ruiz1
1. Hospital Dr. Exequiel González Cortés. Santiago, Chile.
Contenido: El síndrome de insensibilidad androgénica (AIS) es una causa rara de TDS 46XY. Se debe a una mutación en el receptor de andrógenos (RA) causando una resistencia a la acción de éstos. La presentación clínica dependerá del grado de insensibilidad desde fenotipo masculino normal con infertilidad, hasta fenotipo femenino que se presentan con amenorrea 1°, pasando por distintos grados de ambigüedad genital.
Objetivo: describir un caso de AIS parcial causada por nueva mutación del receptor de andrógenos, su presentación clínica, estudio hormonal, anatómico, genético y su manejo.
Caso Clínico: RN de 6 días de vida que es derivado a Endocrinología por TDS, presentándose
con repliegues labio escrotales fusionados, falo de 1,9 x 0,5cm, con cuerpos cavernosos en base, meato urinario próximal, gónadas palpables
de 2 cc en canal inguinal. Antecedente de 2da hija de padres no consanguíneos con media hermana materna con diagnóstico de TDS 46XY
por probable insensibilidad androgénica. En la familia materna hay antecedentes de TSD con mujeres sin descendencia y hombres con ginecomastia, infertilidad, hipospadias y criptorquidia, sin estudio genético. A los 28 días de realiza estudio hormonal: LH: 1,6 mUI/ml, FSH: 2,1mUI/ml Testosterona 85,4 ng/dl, DHT: 68 pg/ml, 17OH Progesterona: 2,17 ng/ml ; a los 2 meses se realiza test de HCG, Testosterona basal
70,5 ng/dl post 634.9 ng/dl. Cariotipo 46X, SRY(+) TSPY(+) DYZ3(+).
Ecografía: gónadas en extremo distal de conductos inguinales que
semejan testículos de 1.08 x 0.37 cm y 1.22 x 0.39 cm, no se observan estructuras müllerianas. Se hace prueba de testosterona con 25 y 50
mg sin obtener ninguna respuesta a nivel genital. Sexo asignado femenino. A los 9 meses uretrocistografía y laparoscopía que describe introito
femenino, seno urogenital, ambas gónadas en canal inguinal que semejan testículos, sin órganos internos femeninos. Biopsia de gónadas:
parénquima testicular inmaduro con cordones serpenteantes. Albugínea libre de cordones. Células con citoplasma vacuolado, con liquidoacidófilo.
Agregados de células germinales de gran tamaño, testículo disgenético. Estudio molecular: análisis por secuenciación automática de exones
y regiones intrónicasflanqueantes del gen AR a partir de ADN genómico: mutación p.Ile907Val. encontrándose la misma mutación homocigota
en media hermana y heterocigota en la madre. Esta mutación no ha sido descrita previamente en pacientes con AIS. Se analizaron los posibles
efectos a nivel de la función proteica de la alteración encontrada mediante la utilización de herramientas informáticas (Polyphen; MutationTaster),
sugiriendo que el cambio aminoacídico afectaría la función de la proteína. Conclusión: se describe un paciente con insensibilidad parcial a
andrógenos causado por una nueva mutación del receptor de andrógenos, son necesarios ensayos in vitro para asignar fehacientemente la
significancia funcional de esta alteración.
Financiamiento: Sin financiamiento.