¿Qué hay de nuevo en el síndrome de ovario poliquístico?
Amanda Ladrón de Guevara H.1,2, Nataly Vantman L.1, Bárbara Echiburú L.1, Daniel Miranda S.2 y Teresa Sir-Petermann1,2
What’s new in polycystic ovary syndrome?
11Laboratorio de Endocrinología y Metabolismo, Facultad de Medicina
Occidente, Universidad de Chile.
2Unidad de Endocrinología-Servicio de Medicina, Hospital San Juan de Dios.
Proyecto SOCHED 2009-05.
Correspondencia:
Amanda Ladrón de Guevara Hernández
Las Palmeras 299, interior Quinta Normal.
Teléfono: 6816693 / 6814676. Fax: 6816693
E-mail: a.ldeguevara@yahoo.com
Recibido: 25 de enero de 2013
Aceptado: 15 de abril de 2013
Polycystic ovary syndrome is recognized as a risk factor for the development of type 2 diabetes and metabolic syndrome. The prevalence of the condition is 6 to 10% in different populations. Its etiology is not well known and there are genetic and epigenetic phenomena involved. Due to its association with insulin resistance, it has been incorporated as another component of Reaven plurimetabolic syndrome. Therefore polycystic ovary syndrome evolved from an ovarian disease to a multisystem disorder and it must be considered a public health problem.
Key words: Polycystic ovary syndrome, polygenic disease, physiopathology.
El síndrome de ovario poliquístico (SOP), es un desorden hiperandrogénico asociado a oligo-ovulación crónica y morfología de ovarios poliquísticos. Es la causa más común de hiperandrogenismo con una incidencia que alcanza 6 a 10% según crietrios descritos en 1990 por el National Institutes of Health en Estados Unidos (NIH) tanto en mujeres adolescentes como adultas1,2. Esta prevalencia aumenta a un 15% si se utilizan los criterios de Rotterdam descritos en el año 20033. Su etiología es incierta y se manifiesta por síntomas y signos variados que afectan a cada mujer en forma particular. Entre ellos destacan las irregularidades menstruales, manifestaciones cutáneas del hiperandrogenismo, infertilidad, obesidad, RI y el aspecto poliquístico de los ovarios en la ultrasonografía.
Inicialmente el SOP fue definido como una patología reproductiva, basada en las características morfológicas de los ovarios tales como: aumento de tamaño, engrosamiento de la túnica albugínea y microquistes múltiples situados periféricamente en la zona subcortical ovárica4. Posteriormente se determinó que el síndrome clínico podía asociarse a ovarios de morfología aparentemente normal y que hasta el 25% de las mujeres sanas podían presentar imágenes ultrasonográficas sugerentes de ovarios poliquísticos sin el síndrome clínico5, por lo que en la actualidad se acepta que las alteraciones morfológicas de los ovarios no son necesarias para su diagnóstico.
Luego aparecen las primeras publicaciones en que se describe la presencia de RI periférica y su expresión a través de una hipersecreción de insulina en estas mujeres6. Se determinó que el estímulo de insulina promueve una mayor secreción de andrógenos por el ovario y las glándulas suprarrenales; aumenta la secreción de LH y además disminuye la síntesis hepática de la SHBG (globulina trasportadora de hormonas sexuales) con lo cual aumenta la fracción libre y actividad biológica de los andrógenos7-9.
Desde entonces, se han publicado un sinnúmero de estudios clínicos y epidemiológicos que describen una fuerte asociación entre el SOP, la obesidad, hígado graso, dislipidemia, diabetes mellitus tipo 2, hipertensión arterial y riesgo cardiovascular10-12.
En la actualidad se ha avanzado notablemente en la fisiopatología de esta enfermedad.
Se ha determinando que tanto el ovario como la glándula suprarrenal participan activamente en la formación e hipersecreción de andrógenos. Se ha identificado además, órganos potenciadores que, aunque no pueden sintetizar andrógenos de novo, pueden amplificar la actividad androgénica a través de la conversión periférica de hormonas débiles a más potentes, gracias a las enzimas esteroidogénicas que poseen. Esta capacidad se ha determinado tanto en el tejido graso como en el folículo pilosebáceo13. Por otro lado, se ha comprobado un aumento en la actividad del sistema nervioso simpático (SNS) en las pacientes con SOP, cuya acción es capaz de estimular la secreción ovárica de andrógenos14. Estos últimos hallazgos se encuentran en plena investigación y nos ayudan a entender esta enfermedad como una entidad cuya fisiopatología es muy compleja y que dista mucho de estar confinada al ovario.
El SOP es una enfermedad endocrino metabólica de compromiso sistémico de alta prevalencia. Por lo tanto, es evidente, según todos estos argumentos, que este síndrome debe considerarse un problema de salud pública que afecta a la mujer más allá de su esfera reproductiva.
1. Definición
La definición del SOP ha sufrido varias modificaciones
desde su primera descripción.
En 1935 Stein y Leventhal lo definieron como hiperandrogenismo de origen ovárico que clínicamente se presentaba en mujeres jóvenes con obesidad, hirsutismo y amenorrea.
Esta definición, que es la más antigua, rescata lo medular de esta patología: la producción ovárica de andrógenos4.
Con el fin de unificar criterios y dado la heterogeneidad del síndrome, en 1990, en la conferencia de consenso del NIH en los Estados Unidos, se lo definió, como la “presencia de hiperandrogenismo asociado a anovulación crónica sin otra causa específica de enfermedad adrenal o hipofisiaria que curse con irregularidades menstruales o exceso de andrógenos”2.
Posteriormente, la Sociedad Europea de Reproducción y Embriología (ESHRE) y la Sociedad Americana de Medicina Reproductiva (ASRM) en una conferencia de consenso realizada en Rotterdam en el año 2003 propuso una nueva definición, en donde la incorporación de la ecografía ginecológica juega un rol relevante en el diagnóstico3.
En este consenso se determinó que con 2 de 3 criterios presentes, puede establecerse el diagnóstico de SOP. De este modo al agrupar los criterios, pueden desprenderse 4 fenotiposdiferentes de esta enfermedad como se observa en la Figura 1.
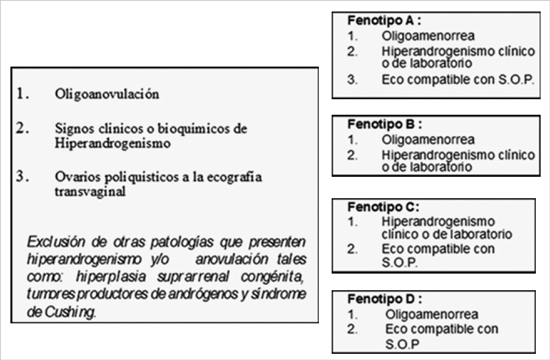
Figura 1. Criterios del consenso de Rotterdam 2003.
De estos, los fenotipos A y B cumplen con los criterios NIH y son los que podríamos considerar un “SOP clásico”. Mientras que el fenotipo C pudiera considerarse un estado de transición o intermedio entre una mujer normal y una SOP clásico15 y el fenotipo D por no tener hiperandrogenismo clínico ni hiperandrogenemia (aumento de andrógenos circulantes) para algunos expertos no debería considerarse dentro del espectro de esta patología asumiendo que por definición el SOP es un trastorno eminentemente hiperandrogénico16. Estos dos últimos fenotipos, son entidades que no cumplen con los criterios NIH y que han suscitado nuevas controversias16,17.
Para mayor claridad en el diagnóstico, en el último workshop de NIH realizado en diciembre de 2012 se recomienda mantener los criterios diagnósticos de Rotterdam y especificar el fenotipo en cada paciente.
Definición de SOP en la adolescencia
No existe claridad para establecer el diagnóstico de esta patología en la adolescencia ya que en esta etapa de la vida, en mujeres sanas, es frecuente el acné, la seborrea así como alteraciones del ciclo menstrual. Se ha descrito en adolescentes sanas que 85% de los ciclos menstruales puede ser anovulatorio el primer año post menarquia, llegando a ser hasta 60% el tercer año posterior a la primera regla1. Con respecto a la ecografía, la morfología SOP puede verse en un 40% de ellas18,19.
Por lo anterior se estableció que para el diagnóstico de SOP es necesario que la mujer adolescente presente los siguientes criterios1:
- Oligo-amenorrea al menos 2 años post menarquia o amenorrea
primaria en mayores de 16 años.
- Ecografía con morfología de ovario poliquístico (MOP)
según criterios de Rotterdam incluyendo tamaño mayor
de 10 cc en uno o dos ovarios los cuales no deben presentar
un folículo dominante.
- Debe demostrarse un hiperandrogenismo de laboratorio y no solamente clínico.
Se entiende por hiperandrogenismo de laboratorio a un aumento de andrógenos plasmáticos (testosterona, androstenediona, dehidroepiandrosterona y/o 17-hidroxiprogestrerona) sobre los rangos normales para una mujer. La determinación de andrógenos circulantes debe realizarse en la fase folicular, es decir, entre el tercer y octavo día desde el inicio de la menstruación.
Es importante recordar que las mujeres con RI sin SOP,
pueden presentar niveles de proteína transportadora de hormonas
sexuales (SHBG) bajas, por acción de la insulina sobre
el tejido hepático. Este fenómeno aumenta la expresión
de andrógenos libres sin existir necesariamente un aumento
en la producción de estas hormonas esteroidales.
Debido a este mecanismo fisiopatológico podemos ver en la clínica mujeres con hiperandrogenismo clínico, índice de andrógenos libres aumentado pero sin un aumento en la producción andrógenos.
En nuestra experiencia, al observar el crecimiento y desarrollo
de las hijas de mujeres con SOP, vemos que este
síndrome se presenta con frecuencia después del 3er año post
menarquía, alcanzando una prevalencia de 33% de acuerdo a
los criterios del Consenso de SOP 20121.
2. SOP enfermedad endocrino-metabólica
El SOP es una disfunción endocrino- metabólica que se
presenta desde etapas tempranas de la vida y condiciona un
mayor riesgo de síndrome metabólico (SM). Esta patología,
en conjunto con la disfunción beta pancreática, que suele
presentarse precozmente en estas pacientes, condiciona la
aparición de intolerancia a la glucosa en un 31,1% y hasta un
7,5% de diabetes mellitus tipo 2. Esto representa un riesgo
relativo para diabetes de 3 a 7 veces mayor comparado con
mujeres de la misma edad y a 2 veces si se compara con mujeres
de igual edad e índice de masa corporal20.
Sumado a estos datos se ha demostrado que las pacientes SOP presentan un riesgo cardiovascular elevado comparado con mujeres de la misma edad e independiente del índice de masa corporal, posiblemente debido al espectro de alteraciones metabólicas descritas en esta enfermedad: alta incidencia de hipertensión arterial, hipercolesterolemia, con altos niveles de LDL (low density lipoprotein), triglicéridos y bajo HDL (high density lipoprotein), acompañado de hígado graso.
Los marcadores de riesgo cardiovascular como proteína C reactiva ultrasensible, homocisteína y mediciones de arteriosclerosis subclínica están aumentados en estas pacientes20.
En las últimas décadas se ha trabajado intensamente en aclarar cuál es el factor común en la etiopatogenia de esta enfermedad que explique las diversas alteraciones endocrino metabólicas. Falta mucho por investigar con respecto a este punto, sin embargo, la insulinoresistencia y la hiperinsulinemia compensatoria es reconocida como el principal factor responsable de la alteración en la producción de andrógenos, la función reproductiva y metabólica de estas pacientes21.
La obesidad por otro lado, sería un elemento amplificador, aumentando la secreción de andrógenos, el deterioro metabólico y reproductivo22.
En el último workshop de NIH (diciembre, 2012) señalan la necesidad de realizar estudios multicéntricos longitudinales que respondan algunas interrogantes actuales como la asociación del síndrome metabólico a un fenotipo específico o el impacto del tratamiento de las anormalidades metabólicas para reducir las complicaciones cardiovasculares y diabetes tipo 2.
3. Fisiopatología del ovario
Producción ovárica de andrógenos
La secreción excesiva de andrógenos, es un pilar fundamental en este síndrome y se caracteriza por una alteración de la biosíntesis en la esteroidogénesis de la célula de la teca.
Clásicamente ubicamos a las células de la teca distribuidas por fuera del folículo y sabemos que son capaces de producir andrógenos de novo a partir del colesterol. Esta acción está aumentada gracias al estímulo de la LH e insulina23,24.
Las células de la teca no sólo están ubicadas alrededor del folículo sino también en el estroma ovárico.
En los últimos años ha tomado mayor relevancia la participación del estroma ovárico como fuente productora de andrógenos. En clínica, su expresión máxima se denomina hipertecosis cuadro que genera un hiperandrogenismo muy marcado e incluso, virilización.
La hipertecosis se caracteriza histológicamente por una hiperplasia estromal de las células de la teca25.
Disfunción de la foliculogénesis
Se ha podido establecer, mediante estudios ultrasonográficos y biopsias ováricas, que las pacientes con SOP presentan un pool de folículos en crecimiento 2 a 3 veces superior que las mujeres sanas. La histología del síndrome de ovario poliquístico se caracteriza por un aumento de folículos preantrales y antrales pequeños y un mayor reclutamiento folicular26. Esta situación se acompaña además de una detención del proceso de selección folicular, lo que explica la ausencia de ovulación27. Por lo tanto, en el síndrome de ovario poliquístico habría mayor reclutamiento y una menor selección, lo que mantiene un aumento del pool de folículos en crecimiento productores de andrógenos.
En los últimos años se ha propuesto que la Hormona Antimülleriana (AMH) podría constituir un marcador sérico que refleje el número de folículos en crecimiento, independiente de gonadotrofinas y por lo tanto de la reserva ovárica en cualquier momento de la vida de la mujer28. Además, recientemente hemos podido establecer que las hijas de mujeres con síndrome de ovario poliquístico tienen niveles significativamente mayores de AMH desde la infancia temprana (2 a 3 meses de vida) hasta la peripubertad, lo que sugiere que estas niñas nacen con una masa de folículos aumentada, lo que podría constituir un eslabón para el desarrollo ulterior de SOP29,30.
4. Fisiopatología suprarrenal
En mujeres normales la secreción adrenal de andrógenos comienza a disminuir alrededor de los 30 años de edad, a diferencia del ovario que lo hace 3 a 4 años post menopausia31.
En mujeres con SOP, se ha observado que la secreción de andrógenos suprarrenales se mantiene incluso después de la menopausia y que contribuye significativamente en el hiperandrogenismo con una capacidad de secretar andrógenos similar a la del ovario32,33.
Se postula que este hiperandrogenismo se debe tanto a un estímulo directo de la insulina sobre la suprarrenal; como a un estímulo indirecto de la insulina al incrementar la producción de factores de crecimiento insulinosímiles a nivelhepático (principalmente IGF-1) que a su vez estimulan la glándula suprarrenal.
En mujeres obesas con SOP, se ha descrito un tercer mecanismo en la génesis del hiperandrogenismo. El tejido adiposo aumentaría la degradación de cortisol a través de las enzimas 11 beta hidroxiesteroide deshidrogenasa tipo 2, 5 alfa reductasa y 5 beta reductasa presentes en el adipocito (Figura 2). Lo cual provocaría un déficit relativo de cortisol plasmático, estimulando de esta forma la secreción hipofisiaria de ACTH la que a su vez, estimularía la esteroidogénesis suprarrenal34,35.
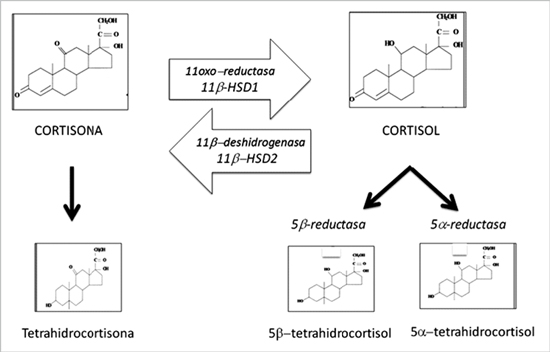
5. Fisiopatología de los mecanismos potenciadores
a. Unidad endocrina pilosebácea y tedido adiposo
Las hormonas esteroidales se caracterizan por ser altamente liposolubles, por lo que penetran fácilmente al tejido adiposo y a la unidad pilosebácea. Estos tejidos son denominados órganos potenciadores de andrógenos.
Si bien el adipocito no tiene la capacidad de producir andrógenos de novo, posee actividad esteroidogénica para transformar hormonas débiles como la dehidroepiandrosterona (DHEA) y la androstenediona a andrógenos más potentes como testosterona y dehidroepiandrosterona36 como se muestra en la Figura 3.
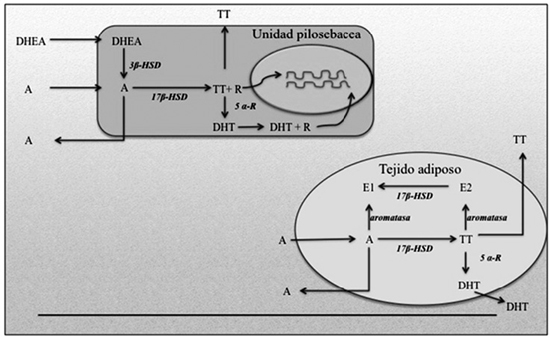
Figura 3. Unidad endocrina: pilosebácea y tejido adiposo. A: Androstenediona; DHEA: dehidroepiandrosterona; TT: testosterona; DHT: dehidrotestosterona; R: receptor esteroidal; 5 a-R: 5 alfa reductasa; 17 b-HSD: 17 hidroxiesteroide deshidrogenasa; E1: estrona; E2: estradiol.
Los principales andrógenos como la testosterona y la androstenediona circulan en un alto porcentaje unidos a la proteína transportadora de hormonas sexuales (SHBG). Esto evita que dichas hormonas se encuentren biodisponibles para ejercer su acción en los órganos potenciadores actuando como un verdadero reservorio plasmático que se mantiene inactivo mientras se encuentre ligado a esta proteína. Las mujeres con SOP y RI presentan bajos niveles plasmáticos de SHBG por efecto de la insulina sobre el hígado15,21, por lo que poseen mayores concentraciones de andrógenos libres circulantes susceptibles de actuar a nivel del tejido adiposo y unidad pilosebácea.
Como mencionamos anteriormente, el tejido adiposo participa en el metabolismo periférico del cortisol por lo tanto, en mujeres con un alto porcentaje de grasa corporal esto puede llegar a activar el eje corticotropo34,35.
b. Sistema Nervioso Simpático e Hiperandrogenismo ovárico
Los ovarios de mujeres con SOP, presentan una gran densidad de fibras nerviosas catecolaminérgicas, con aumento de actividad del SNS local37.
Se ha planteado que el incremento de actividad simpática puede contribuir al aumento de secreción de andrógenos ováricos38. Se ha demostrado en modelos de ratas transgénicas y en mujeres con SOP, un aumento del factor de crecimiento nervioso (NGF) en tejido ovárico, marcador de la actividad del SNS la cual se relaciona con el grado de producción de testosterona y severidad del SOP39,40.
Debido a estos hallazgos, existen grupos de investigación que incentivan el ejercicio, especialmente el yoga para atenuar la actividad simpática.
Otros grupos sugieren la electroacupuntura como método disruptor de la inervación ovárica41,42.
6. Morfología SOP en ecografía
La morfología tipo SOP de ovario poliquístico (MOP), según criterios de Rotterdam puede observarse entre un 17 a 33% de mujeres adultas sanas21 y hasta un 40% en adolescentes sanas18. Por lo anterior, este hallazgo aislado, no constituye diagnóstico de síndrome de ovario poliquístico.
Por otro lado, la presencia de MOP se ha descrito como marcador de reserva ovárica y de envejecimiento ovárico más tardío43.
En mujeres adultas existe debate sobre significado clínico de estos hallazgos ecográficos. Es así como, en pacientes con hipogonadismo hipogonadotrópico y pacientes epilépticas en tratamiento con acido valproico se ha observado la presencia de MOP en la ecografía y una hipersecreción de LH al estímulo con análogos de GnRH44,45. Lo que sugiere que este aspecto ecográfico no es patognomónico del SOP. Lo mismo es válido para la etapa postmenárquica inicial, en que niñas sanas también presentan este tipo de imagen ecográfica19.
En suma, los hallazgos característicos de MOP en la ecografía, no constituyen un diagnóstico por sí solos, pero es un rasgo fenotípico que debe controlarse21.
7. Hipertecosis
Nos referiremos a esta patología que a pesar de ser poco
frecuente, en los últimos años hemos observado un aumento
de casos en el policlínico de nuestro laboratorio.
La hipertecosis se caracteriza por una hiperplasia estromal de células de la teca, que pueden distribuirse en forma aislada, como pequeños nidos o incluso, formar nódulos. La hiperinsulinemia es la base de esta patología. El estímulo insulínico sostenido sobre estas células, produciría la hiperplasia.
Clínicamente se caracteriza por hiperandrogenismo severo que puede llegar a la virilización, acompañado por un cuadro metabólico muy acentuado25,46.
Suele acompañarse de hipertensión arterial, alteraciones del metabolismo de hidratos de carbono, dislipidemia y obesidad. En la literatura hay pocos casos descritos y la mayoría ocurren en la post menopausia46,47. Hemos observado un número creciente de esta entidad desde la etapa reproductiva temprana hasta la post menopausia posiblemente debido a la alta prevalencia de síndrome metabólico e insulinoresistencia que existe en nuestro país48.
De acuerdo a los casos que hemos observado, el perfil hormonal de estás pacientes se caraceriza por elevados niveles de testosterona (mayor a 1 ng/ml), concomitante con el aumento de otros andrógenos de origen ovárico como androstenediona y 17 OH progesterona lo cual hace indispensable descartar tumores virilizantes o hiperplasia suprarrenal congénita como diagnósticos diferenciales.
Las características ecográficas son variables. Lo más frecuente es encontrar un ovario normal o con un volumen aumentado a expensas del estroma ovárico o una MOP clásica, sugiriendo que la interpretación ecográfica de la hipertecosis es compleja49,50.
Finalmente el diagnóstico de esta patología es histológico por lo que sólo puede realizarse en forma certera a través de una biopsia ovárica, siendo su tratamiento quirúrgico.
A través de esta pequeña revisión deseamos demostrar que el nombre “síndrome de ovario poliquístico” no refleja en esencia lo que esta entidad representa debido a que es una patología donde se entrecruzan alteraciones de numerosos órganos y sistemas, comprometiendo varios aspectos de la salud en la mujer y no solamente de su esfera reproductiva.
Agradecimientos
Deseamos agradecer a la Sociedad Chilena de Endocrinología
y Metabolismo (proyecto SOCHED 2009-05) y Proyecto
Fondecyt 1110864 y a los miembros académicos y no
académicos del Laboratorio de Endocrinología y Metabolismo
de la Facultad de Medicina Occidente de la Universidad
de Chile por su colaboración.
Referencias bibliográficas
- Fauser B, Tarlatzis B, Rebar R, Legro R. 2012. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): the Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil Steril 97: 28-38.
- Carmina E. 2004. Diagnosis of polycystic ovary syndrome: from NIH criteria to ESHRE-ASRM guidelines. Minerva Ginecol 56: 1-6.
- Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group 2004. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril 81: 19-25.
- Stein IF, Leventhal ML. 1935. Amenorrhea associated with bilateral polycystic ovaries. Am J Obstet Gynecol 29: 181-191.
- Polson DW, Adams J, Wadsworth J, et al. 1988. Polycystic ovaries: a common finding in normal woman. Lancet 1: 870-878.
- Nestler JE, Powers LP, Matt DW, et al. 1991. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. Clin Endocrinol Metab 72: 83-89.
- Dunaif A, Segal KR, Futterweit W, et al. 1989. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 38: 1165-1174.
- Adashi EY, Hsueh AJ, Yen SS. 1981. Insulin enhancement of luteinizing hormone and follicle-stimulating hormone release by cultured pituitary cells. Endocrinology 108: 1441-1449.
- Homburg R. 2009. Androgen circle of polycystic ovary syndrome. Hum Reprod 24: 1548-1555.
- Legro RS, Kunselman AR, Dodson WC, et al. 1999. Prevalence and predictors of risk for Type II diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab 84: 165-169.
- Cerda C, Pérez-Ayuso RM, Riquelme A, et al. 2007. Nonalcoholic fatty liver disease in women with polycystic ovary syndrome. J Hepatol 47: 412-417.
- Moran LJ, Misso ML, Wild RA, Norman RJ. 2010. Impaired glucose tolerance, type 2 diabetes and metabolic syndrome in polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod Update 16: 347-363.
- Pasquali R, Casimirri F. 1993. The impact of obesity on hyperandrogenism and polycystic ovary syndrome in premenopausal women. Clinical Endocrinology (Oxford), 39: 1-16.
- Heider U, Pedal I, Spanel-Borowski K. 2001. Increase in nerve fibers and loss of mast cells in polycystic and postmenopausal ovaries. Fertility Sterility 75: 1141-1147.
- Diamanti-Kandarakis E, Dunaif A. 2012. Insulin Resistance and the Polycystic Ovary Syndrome Revisited: An Update on Mechanisms and Implications. Endocrine Reviews. First published ahead of print October 12, 2012 as doi:10.1210/er.2011-1034.
- Azziz R. 2006. Controversy in clinical endocrinology: diagnosis of polycystic ovarian syndrome: the Rotterdam criteria are premature. J Clin Endocrinol Metab 91: 781-785.
- Franks S. 2006. Controversy in clinical endocrinology: diagnosis of polycystic ovarian syndrome: in defense of the Rotterdam criteria. J Clin Endocrinol Metab 91: 786-789.
- Codner E, Villarroel C, Eyzaguirre FC, et al. 2011. Polycystic ovarian morphology in postmenarchal adolescents. Fertil Steril 95: 702-706.
- Villarroel C, Merino PM, López P, et al. 2011. Polycystic ovarian morphology in adolescents with regular menstrual cycles is associated with elevated anti-Mullerian hormone. Hum Reprod 26: 2861-2868.
- Harpal R, Bee T, Martin W, Weickert L, John N, Naveed S, et al. 2012. Cardiometabolic Aspects of the Polycystic Ovary Syndrome. Endocrine Reviews 33: 812-841.
- Pasquali R, Stener-Victorin E, Yildiz B, Duleba A, Hoeger K, Mason H. 2011. PCOS Forum: research in polycystic ovary syndrome today and tomorrow. Clinical Endocrinology 74: 424-433.
- Escobar-Morreale HF, Botella-Carretero JI, Álvarez-Blasco F, et al. 2005. The polycystic ovary syndrome associated with morbid obesity may resolve after weight loss induced by bariatric surgery. Journal of Clinical Endocrinology & Metabolism 90: 6364-6369.
- Gilling-Smith C, Willis DS, Beard RW, Franks S. 1994. Hypersecretion of androstenedione by isolated thecal cells from polycystic ovaries. J Clin Endocrinol Metab 79: 1158-1165.
- Sir-Petermann T, Cartes A, Maliqueo M, et al. 2004. Patterns of hormonal response to the GnRH agonist leuprolide in brothers of women with polycystic ovary syndrome: a pilot study. Hum Reprod 19: 2742-2747.
- Hughesdon PE 1982. Morphology and morphogenesis of the Stein- Leventhal ovary and of so-called “hyperthecosis”. Obstet Gynecol Surv 37: 59-77.
- Webber LJ, Stubbs S, Stark J, Trew GH, Margara R, Hardy K, et al. 2003. Formation and early development of follicles in the polycystic ovary. Lancet 362: 1017-1021.
- Jonard S, Dewailly D. 2004. The follicular excess in polycystic ovaries, due to intraovarian hyperandrogenism, may be the main culprit for the follicular arrest. Human Reprod 10: 107-117.
- Sowers MR, Eyvazzadeh AD, McConnell D, Yosef M, Jannausch ML, Zhang D, et al. 2008. Anti-Mullerian Hormone and Inhibin B in the Definition of Ovarian Aging and the Menopause Transition. J Clin Endocrinol Metab 93: 3478-3483.
- Sir-Petermann T, Codner E, Maliqueo M, et al. 2006. Increased anti-Müllerian hormone serum concentrations in prepubertal daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab 91: 3105-3109.
- Crisosto N, Codner E, Maliqueo M, et al. 2007. Anti-Müllerian hormone levels in peripubertal daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab 92: 2739- 2743.
- Lasley BL, Santoro N, Randolf JF, et al. 2002. The relationship of circulating dehydroepiandrosterone, testosterone, and estradiol to stages of the menopausal transition and ethnicity. Journal of Clinical Endocrinology & Metabolism 87: 3760-3767.
- Piltonen T, Koivunen R, Ruokonen A, et al. 2003. Ovarian age-related responsiveness to human chorionic gonadotropin. Journal of Clinical Endocrinology & Metabolism 88: 3327-3332.
- Puurunen J, Piltonen T, Jaakkola P, et al. 2009. Adrenal androgen production capacity remains high up to menopause in women with polycystic ovary syndrome. Journal of Clinical Endocrinology & Metabolism 94: 1973-1978.
- Stewart PM, Shackleton CH, Beastal, GH, et al. 1990. 5 alpha-reductase activity in polycystic ovary syndrome. Lancet 335: 431-433.
- Gambineri A, Forlani G, Munarini A, et al. 2009. Increased clearance of cortisol by 5beta-reductase in a subgroup of women with adrenal hyperandrogenism in polycystic ovary syndrome. Journal of Endocrinological Investigation 32: 210-218.
- Barber M, McCarthy I, Wass A, Franks S. 2006. Obesity and polycystic ovary syndrome. Clin Endocrinol (Oxf) 65: 137-45.
- Lara H, Dissen A, Leyton V, Paredes A, Fuenzalida H, Fiedler L, et al. 2000. An increased intraovarian synthesis of nerve growth factor and its low affinity receptor is a principal component of steroid-induced polycystic ovary in the rat. Endocrinology 141: 1059-1072.
- Heider U, Pedal I, Spanel-Borowski K. 2001. Increase in nerve fibers and loss of mast cells in polycystic and postmenopausal ovaries. Fertility Sterility 75: 1141-1147.
- Dissen GA, García-Ruda C, Paredes A, et al. 2009. Excessive ovarian production of nerve growth factor facilitates development of cystic ovarian morphology in mice and is a feature of polycystic ovarian syndrome in humans. Endocrinology 150: 2906-2914.
- Sverrisdottir YB, Mogren T, Kataoka J, et al. 2008. Is polycystic ovary syndrome associated with high sympathetic nerve activity and size at birth? American Journal of Physiology. Endocrinology and Metabolism 294: E576-E581.
- Jedel E, Labrie F, Oden A, et al. 2011. Impact of electroacupuncture and physical exercise on hyperandrogenism and oligo/amenorrhoea in women with polycystic ovary syndrome: a randomized controlled trial. Am J Physiol Endocrinol Metab 300: E37-45.
- Stener-Victorin E, Jedel E, Janson PO, et al. 2009. Low-frequency electroacupuncture and physical exercise decrease high muscle sympathetic nerve activity in polycystic ovary syndrome. Am J Physiol Regul Integr Comp Physiol 297: R387-395.
- Mulders AG, Laven JS, Eijkemans MJ, et al. 2004. Changes in anti-Mullerian hormone serum concentrations over time suggest delayed ovarian ageing in normogonadotrophic anovulatory infertility. Human Reproduction 19: 2036-2042.
- Schachter M, Balen AH, Patel A, et al. 1996. Hypogonadotropic patients with ultrasonographically detected polycystic ovaries: endocrine response to pulsatile gonadotropin-releasing hormone. Gynecological Endocrinology 10: 327-335.
- Isojarvi JI, Laatikainen TJ, Pakarinen AJ, et al. 1993. Polycystic ovaries and hyperandrogenism in women taking valproate for epilepsy. New England Journal of Medicine 329: 1383-1388.
- Barth JH, Jenkins M, Belchetz PE. 1997. Ovarian hyperthecosis, diabetes and hirsuties in post-menopausal women. Clinical Endocrinology (Oxford) 46: 123-128.
- Goldman J, Kapadia L. 1991. Virilization in a postmenopausal woman due to ovarian stromal hyperthecosis. Postgrad Med J 67: 304-306.
- Ministerio de Salud Pública de Chile. Encuesta Nacional de Salud, 2009-2010.
- Brown DL, Henrichsen TL, Clayton AC, et al. 2009. Ovarian Stromal Hyperthecosis: Sonographic Features and Histologic Associations. J Ultrasound Med 28: 587-593.
- Rousset P, Gompel A, Christin-Maitre S, et al. 2008. Ovarian hyperthecosis on grayscale and color Doppler ultrasound. Ultrasound Obstet Gynecol 32: 694-699.