Hiperplasia suprarrenal no clásica, características clínicas y genéticas
Alejandro Martínez A.1, Karime Rumie C.1, Helena Poggi M.2, Hernán Garcia B.3, Verónica Mericq G.4, Eugenio Arteaga U.5, José Manuel López M.5, Claudia Campusano M.5, Gilberto González V.5, Carlos Fardella B.5, Paulina Villaseca D.5, Andreina Cattani O.1
Clinical and genetic features of non classical adrenal hyperplasia
1Departamento de Pediatría, Facultad de Medicina de la Pontificia Universidad Católica de Chile.
2Laboratorio de Biología Molecular, Facultad de Medicina de la Pontificia Universidad Católica de Chile.
3Clínica Santa María.
4Clínica Las Condes.
5Departamento de Endocrinología, Facultad de Medicina de la Pontificia Universidad Católica.
Correspondencia a: Dra. Andreina Cattani Ortega. Departamento de Pediatría Pontificia Universidad Católica de Chile Lira 85, Santiago. Código postal: 833-0074, Santiago, Chile. e-mail: acattani@med.puc.cl
Recibido el 31 de octubre, 2007.
Aceptado el 06 de marzo, 2008.
Background: The non classical form of congenital adrenal hyperplasia (NCAH) is increasingly recognized in hyperandrogenic patients, with variable phenotypic expression. Aim: To determine the clinical, hormonal, and genetic characteristics of a group of patients with NCAH. Patients and methods: The medical records of 57 NCAH patients were retrospectively reviewed. The diagnosis was established by basal or post-ACTH-stimulation 17-hydroxyprogesterone (17-OHP) levels >7 ng/mL and > 15 ng/mL, respectively. Patients with post-ACTH 17-OHP levels between 10-15 ng/mL, and with one identified allele o without genetic tests, were considered as heterozygous. Genotyping for 10 mutations was performed by PCR. Results: The average age of diagnosis was 12.4± 0.9 years. Six patients were male. Pubarche and hirsutism were the clinical signs more frequently described in patients below 10 years of age (25/29) and over 10 years of age (11/24), respectively. A basal 17-OHP > 7 ng/mL was observed in 36 patients; the post ACTH 17-OHP was between 10-15 and > 15 ng/mL in 5 and 17 patients, respectively. Genotype analyses were performed in 38 patients. V281L was carried on approximately 68.4% of all alleles and 29% of patients carried severe mutations. Only one of five possible carrier patients, was diagnosed as NCAH after the genetic test (V281L/ In2splice). Conclusions: Males with NCAH were apparently sub-diagnosed. Pubarche and hirsutism were the more frequently reported signs. The genetic test is complementary in the diagnosis of NCAH. One third of the patients carried a classic mutation and could have an increased risk to have siblings with Classical CAH.
La Hiperplasia Suprarrenal Congénita (HSC) se produce por una variedad de errores de la esteroidogénesis suprarrenal, derivados de defectos en una de las cinco enzimas que participan en la producción de cortisol. En un 95% de los casos, la HSC obedece a deficiencia de la 21-hidroxilasa (21-OH); la 21-hidroxilación suprarrenal es catalizada por el citocromo P450c21 y codificada por el gen CYP21B. La mayoría de las mutaciones en este gen son producidas por conversión génica, en la cual se transfieren secuencias inactivas presentes en el seudogen, o también puede ocurrir la deleción parcial o total de él. Esto origina pérdida o disminución de la actividad de la 21-OH, que se traduce en disminución de la síntesis de cortisol y aldosterona asociado a aumento de la producción de andrógenos suprarrenales.
Las formas clínicas de HSC dependen del grado de compromiso de la actividad enzimática. La forma clásica o grave, tanto en la variedad perdedora de sal como en la virilizante simple, se asocia a una actividad enzimática prácticamente nula; en el sexo femenino produce grave virilización desde la vida intrauterina e insuficiencia suprarrenal. Si la enzima conserva entre un 20 a 60% de su actividad, se presentará como una forma moderada o No Clásica (HSC-NC)1.
La HSC-NC es uno de los desórdenes autonómicos recesivos más frecuentes en el ser humano, con una prevalencia de 1:100 sujetos en población blanca no judía y hasta en un 10% en mujeres caucásicas con hiperandrogenismo. Sin embargo, son pocos los estudios que logran reunir un número importante de pacientes2, posiblemente por un bajo índice de sospecha considerando la amplia variabilidad fenotípica y que en el varón puede ser poco sintomática.
Estos pacientes no tienen deficiencia clínica de cortisol ni aldosterona y el hiperandrogenismo suele manifestarse en forma tardía, en la infancia, pubertad o adultez3. Durante la infancia pueden existir síntomas o signos tales como: talla alta, aceleración de la edad ósea, pubarquia prematura4, pubertad precoz; durante la adolescencia suelen consultar por acné grave5, hirsutismo6, desórdenes menstruales; en la vida adulta por oligomenorrea, Síndrome de Ovarios Poliquísticos, e incluso, infertilidad. Estudios en familiares de afectados con una forma clásica o en tamizajes de HSC a nivel poblacional demuestran que existen sujetos con HSC-NC, que no presentan manifestaciones clínicas, lo que se conoce como forma críptica de HSR-NC7.
La mayoría de los pacientes son heterocigotos compuestos y la forma de presentación clínica se correlacionará con la actividad enzimática del alelo menos afectado3-6.
Nuestros objetivos fueron conocer las características clínicas que motivaron la consulta médica, las concentraciones basales y post estímulo con ACTH de 17-OHP y su relación con el estudio genético de un grupo de pacientes chilenos con HSC-NC.
Pacientes y Métodos
El estudio es de carácter descriptivo y retrospectivo. Todos los pacientes fueron derivados con diagnóstico bioquímico y/o molecular de HSC-NC y controlados en su mayoría en el policlínico de endocrinología pediátrica o de adultos de la Pontificia Universidad Católica de Chile.
El diagnóstico se realizó basado en los síntomas y signos sugerentes de hiperandrogenismo, niveles plasmáticos basales de 17-OHP > 7 ng/mL y/o post estímulo de ACTH (0,25 mg) > 10 ng/mL. Aquellos pacientes con 17-OHP post estímulo de ACTH entre 10 y 15 ng/mL debían tener estudio genético confirmatorio. Los casos con sólo un alelo identificado serán analizados como sub-grupo ante la eventualidad de ser heterocigotos (“portadores”).
En algunos pacientes se determinó la concentración plasmáticas de testosterona total (n= 37) y DHEA-S (n=35).
Para el analisis clinico los pacientes fueron estratificados en dos grupos, Grupo 1: < 10 anos y Grupo 2 ≥ 10 años. En el Grupo 1 se consigno el motivo de consulta: talla alta, pubarquia prematura o pubertad precoz; acne y/o sudoracion apocrina antes de los 6 años de edad. Se determino la edad osea con radiografia de carpo, según el metodo de Greulich and Pyle8. Se calculo la talla diana. En el Grupo 2 se consigno: amenorrea primaria, definida como ausencia de regla despues de los 15 anos; amenorrea secundaria, entendida como ausencia de regla en los tres ultimos meses y oligoamenorrea, cuando habia menos de nueve reglas por ano. La infertilidad fue obtenida como dato anamnestico. En el examen fisico se considero como hirsutismo un puntaje de Ferriman mayor de 109; se consigno la presencia de alopecia y clitorimegalia.
Ensayos hormonales
Los exámenes hormonales se realizaron después de ayuno nocturno y antes de las 9 AM. En las mujeres post-menárquicas los exámenes fueron realizados en la fase folicular del ciclo. Se determinó 17-OHP en condición basal y después de 60 minutos de una dosis de 0,25 mg de ACTH (Cortrosyn, 0,25 mg iv; Alliance Pharmaceutical, Whitshire, UK).
La 17-OHP fue determinada por RIA (Diagnostic Product Corp.,
Los Angeles, CA). El limite de deteccion fue 0,1 ng/mL y el coeficiente de variacion 8%. La testosterona fue determinada por ICMA (Modular Analitics E170, ROCHE) con un CV interensayo de 7,4% para una concentracion de 17 ng/dL, 2,2% para 6 ng/dL y de 1,7% para 118 ng/mL, respectivamente. La DHEA-S fue determinada por ICMA (Immulite 2002, DPC) con un CV interensayo de 9,8% para 1,6 μg/mL y menor de 7% para niveles entre 2,64 y 6,59 μg/mL.
Analisis genetico
El ADN genómico se extrajo a partir de linfocitos de sangre periférica utilizando métodos convencionales10. Por PCR aleloespecífico, de acuerdo a lo descrito por Fardella y col.11,-13, se estudiaron 10 mutaciones puntuales descritas como las más frecuentes en distintas poblaciones (todas derivadas del seudogén a excepción de la producida por Pro453Ser). Las mutaciones asociadas a forma clásica son: Pro30Leu, In2splice, E3del8bp, Ile172Asn, ClusterE6, Leu306insT, Gln318stop, Arg356Trp, y la deleción o macroconversión (Del/MC). Las mutaciones asociadas a forma no clásica son: Val281Leu, Pro30Leu, y Pro- 453Ser. Usando esta metodología, el porcentaje de mutaciones identificadas en nuestro laboratorio, varía entre un 73,7% a 77% en pacientes con HSC-Clásica13,14 y 75% en pacientes con HSCNC14.
Análisis Estadístico
Los resultados se presentan como promedio y desviación estándar. Para evaluar la posible asociación entre dos variables sin distribución normal, se usó la Prueba de Spearman y para determinar si había diferencia entre dos variables dependientes, la Prueba de Wilcoxon. Se consideró estadísticamente significativo un p <0,05. El análisis estadístico se realizó utilizando el programa computacional SPSS 10,0 para Windows.
Resultados
Cincuenta y siete pacientes fueron derivados con diagnóstico presuntivo de HSC-NC. Seis pacientes fueron de sexo masculino (10,5%). La edad al momento del diagnóstico fue 12,4± 0,9 años (dispersión: 2,6 a 29 años).
Del total de 57 pacientes, 36 tenían 17-OHP basal >7 ng/mL, con promedio de 28,9± 31 (dispersión: 7,2 a 170 ng/mL). En los 21 individuos restantes, la 17-OHP post estímulo con ACTH fue 29,7± 16 ng/mL (dispersión: 10 a 62,7 ng/mL); en 17 personas, ella fue mayor de 15 ng/mL y en 5 pacientes entre 10 y 15 ng/mL. En éstos últimos sujetos, el estudio genético detectó a un paciente heterocigoto compuesto para mutación Val281Leu/ In2splice (Paciente #20 en Fig. 1) y a 3 pacientes con Val281Leu /No Detectado (Pacientes #1, #2, #5 en Fig.1); el paciente #18 no tenía estudio genético Los últimos cuatro sujetos se consideraron como probables portadores y se analizan en forma independiente. En un paciente con HSC-NC (1,9%), el nivel de 17-OHP basal fue < 2 ng/mL. La Figura 1 muestra la asociación entre la 17-OHP basal y la post estímulo con ACTH.
En el Grupo 1 (n= 29; mujeres= 24) el motivo de la consulta médica fue: pubarquia prematura (25/29), sudor apocrino antes de los 6 años (9/29), pubertad precoz central (6/29), talla alta (3/29) y acné grave (2/29). En el Grupo 2 (n=24; mujeres=23) consultaron por hirsutismo (11/23), con un puntaje de Ferriman entre 10 a 29 puntos, oligoamenorrea (5/24), acné grave (5/24). Un varón consultó por pubertad temprana.
La edad ósea (EO) estuvo disponible en 26/53 sujetos, la relación edad ósea/edad cronológica fue 1,12± 0,19 (dispersión: 0,86 a 1,64). La EO fue concordante con la edad cronológica en el 65,4% de los sujetos estudiados. La talla final estuvo disponible en 15 sujetos (13 mujeres y 2 hombres). No se observó diferencia entre la talla diana y la talla final alcanzada; en el caso de las mujeres fue 158,6 ± 1,9 /vs. 158 ± 1,9 cm, respectivamente (Wilcoxon; p= 0,625); y en el caso de los hombres 167 ± 9,7 /vs. 171 ± 0,5 cm, respectivamente (Wilcoxon; p= 0,7). Además, no encontramos asociación entre la talla final y la edad al momento del diagnóstico (Spearman, p= 0,684).
En el Grupo 1, los niveles plasmáticos de testosterona fueron evaluados en 19 de 29 pacientes; estando dentro del valor de referencia (VR) para la edad en 57,9% de los sujetos; con un promedio de 35,4 ± 23,8 ng/dL (dispersión: 10 a 96 ng/dL); tres sujetos tenían testosterona total <10 ng/dL; de ellos, dos eran de sexo masculino (con niveles de 17-OHP basal de 11 y 13 ng/mL). En el Grupo 2 las mujeres fueron evaluadas con testosterona total en 18 de 23 sujetos; en ellas el promedio de testosterona total 91 ± 49 ng/dL (dispersión: 19 a 203 ng/dL); cuatro pacientes tenía valores menores de 60 ng/dL, con niveles de 17-OHP basal que fluctuaban entre 4,6 a 31,0 ng/mL.
En el Grupo 1 las concentraciones de DHEA-S se determinaron en 23 de 29 pacientes; el promedio fue 0,92 } 0,53 μg/mL (dispersion: 0,25 a 2,0 μg/mL). En el Grupo 2 la DHEA-S se determino en 12 de 23 mujeres y el promedio fue 3,2 } 2,1 μg/mL (dispersion: 0,16 a 7,3 μg/mL). Tenian niveles plasmaticos dentro del valor de referencia normal un 78,3% del Grupo 1 (VR; 0,3- 1,29 ug/mL) y 50% del Grupo 2 (VR; 0,34-2,55 ug/mL).
Del total de pacientes estudiados, el estudio genetico estuvo disponible en 38 de 57 (66,7%) sujetos y se muestra en las Tablas 1 y 2.
De los cuatro sujetos considerados como “probables portadores”, en tres sólo se identificó un alelo afectado (Pacientes #1; #2; #5, Val281Leu /ND; Fig.1); un paciente no tuvo estudio genético (#18). El promedio de 17-OHP basal fue 2,8± 2,1 ng/mL (dispersión: 1,3 a 5,9 ng/mL) y post ACTH 11,6± 1,4 ng/mL (dispersión: 10 a 13,3 ng/mL). La testosterona total, fue 10 y 44 ng/dL en los niños <10 años; y 65 y 110 ng/dL en los sujetos >10 años.
Se realizó estudio familiar en 3 de 53 de los casos índices, detectándose cinco pacientes con HSC-NC, todos con 17-OHP post ACTH >15 ng/mL; tres de los cinco estaban asintomáticos (crípticos). Durante el seguimiento, todos desarrollaron hiperandrogenismo sintomático.
Discusión
Este es el primer artículo con un importante número de pacientes chilenos con Hiperplasia Suprarrenal Congénita No Clásica en quienes evaluamos las características clínicas, bioquímicas y genéticas. Destacamos que los hombres están subdiagnosticados, que la pubarquia y el hirsutismo fueron los signos clínicos más frecuentemente descritos en niños y adolescentes, respectivamente; además, un nivel basal de 17-OHP <2 ng/mL no descarta HSC-NC en pacientes con alta sospecha clínica, de modo que en ellos se debe realizar la prueba de estímulo con ACTH. Uno de los resultados más importantes a considerar es que un tercio de los pacientes eran portadores de mutaciones asociadas a la forma clásica de HSC, quienes tendrían riesgo de tener hijos con HSC clásica.
La HSC-NC origina grados variables de hiperandrogenismo que se pueden manifestar desde la niñez hasta la vida adulta15; nuestros pacientes presentaron amplia dispersión de edad al momento del diagnóstico (2,9 a 29 años) y diversos grados de hiperandrogensimo. El seguimiento de pacientes crípticos pone en evidencia el carácter evolutivo del fenotipo de la HSC-NC16.
Si consideramos que la HSC-NC es una patología autosómica recesiva y que afecta a hombres y mujeres por igual, el escaso número de pacientes de sexo masculino en nuestra casuística (10,5%) pone en evidencia el sub-diagnóstico en los varones.
La manifestación clínica más frecuente en el grupo de menores de 10 años fue la pubarquia prematura (25/29), seguida de sudoración apocrina. Importante de resaltar es que en 3 de 29 pacientes, la única manifestación que motivó el estudio fue una estatura inapropiadamente alta en relación a su talla diana. La aceleración de la velocidad de crecimiento, aun sin signos de virilización, debe hacer plantear este diagnóstico3,22-24.
La edad ósea fue concordante con la edad cronológica en el 65,4% de los pacientes. New y col2, observaron que en niños diagnosticados antes de los 12 años sólo un 69% tenía avance de la edad ósea.
En los 15 sujetos que alcanzaron talla final, ésta fue concordante con su carga genética, no importando la edad del diagnóstico, posiblemente porque el grado de hiperandrogenismo fue leve. Sin embargo, otros autores han demostrado que el grado de avance en la edad ósea puede determinar deterioro de la talla final en esta variedad de hiperplasia suprarrenal17-19.
Las mujeres post-menárquicas con HSC-NC pueden tener disfunción menstrual como amenorrea, anovulación, oligoamenorrea20 y/o infertilidad21; esto ha sido atribuido a una disrupción de la secreción de gonadotrofinas por retroalimentación continua ejercida por los esteroides de origen suprarrenal en el eje hipotálamo-hipofisiario22. Además, pueden presentar hirsutismo como único signo de HSC-NC23. En nuestro estudio, el hirsutismo fue el motivo de consulta más importante en las mayores de 10 años y fue habitualmente moderado.
En relación a los exámenes diagnósticos, la prueba de estímulo con ACTH constituye una herramienta fundamental. En la Figura 1 se observa que si bien la asociación entre la 17-OHP basal y la posterior al estímulo con ACTH fue significativa, existe gran dispersión. Cabe destacar que un paciente con HSC-NC incluso tenía 17-OHP basal <2 ng/mL. Por este, motivo si existe sospecha fundada de hiperandrogenismo, se debe efectuar la prueba de estímulo con ACTH.
New y col24 sugirieron para el diagnóstico de HSR-NC un valor de corte de 17-OHP post ACTH mayor a 10 ng/mL; este valor corresponde al límite superior de los sujetos heterocigotos con deficiencia de 21OH antes que el estudio genético estuviera ampliamente disponible. Posteriormente, algunos investigadores han puesto en duda este nivel de corte9,25 porque entre 10 a 15 ng/mL hay pacientes que pueden ser heterocigotos. En nuestro estudio, cinco pacientes tenían 17-OHP post ACTH entre 10 y 15 ng/mL; de ellos uno era heterocigoto compuesto, confirmándose la HSC-NC. En los otros cuatro, uno no tenía estudio molecular y en tres pacientes sólo se identificó un alelo afectado, de modo que hasta que no se realice la secuenciación del gen CYP21, no se puede afirmar el diagnóstico de HSC-NC. En los sujetos considerados como portadores, las manifestaciones de hiperandrogenismo podrían estar agravadas por aumento de la sensibilidad del receptor de andrógenos o de la producción de andrógenos a nivel ovárico.
Una vez definida las características clínicas y hormonales, el estudio genético es importante, no sólo para documentar el diagnóstico, sino para realizar un adecuado consejo genético. En nuestro caso se realizó en 38 casos índices e identificamos 85% de los alelos estudiados. El porcentaje de identificación actual fue mayor al que previamente conocíamos14. La mutación más frecuente fue Val281Leu que se encontró en 52/76 alelos, lo que representa un 68,4%, hecho concordante con lo reportado en HSC-NC en diferentes grupos étnicos21, y mayor a lo reportado en Brasil por el grupo de Mendoca y col.26, quienes ubicaron una mutación en el 73% de los alelos estudiados, con una frecuencia para V281L de 42%. Hay que destacar que en Chile, a la fecha, no se realiza la secuenciación completa del CYP21 sin fines de investigación; en este trabajo el hecho de que se estudiaron solo las 10 mutaciones más frecuentes explica el porcentaje de alelos no identificados.
En pacientes con HSC-NC, la presencia de alelos afectados con mutaciones graves varía entre 27% a 76%27. En nuestra casuística, en 11 de 38 pacientes (29%) uno de sus alelos estaba afectado por una mutación asociada a la HSC-Clásica. Asumiendo la prevalencia de heterocigocidad de mutaciones graves en población general (1:60), el cálculo de probabilidad de tener un niño afectado con HSC clásica, entre pacientes con HSC-NC es de 1:4805,6, probabilidad significativamente mayor que la de la población general (1:12,000-23,000). Por otra parte, considerando que la prevalencia de portadores de mutaciones moderadas en población general es de 1:161,28, la probabilidad que una mujer con HSC-NC tenga un hijo con HSC-NC es muy alta, aproximadamente 1:32, particularmente si se considera la prevalencia de HSC-NC en población general (1:400). Por este motivo, el estudio molecular es muy importante no sólo para apoyar el diagnóstico, sino para realizar un adecuado consejo genético. Esto plantea la necesidad de realizar el estudio genético de la pareja, idealmente secuenciado el gen CYP21. En conclusión, la HSC-NC constituye un desorden cuyas manifestaciones son variables y progresivas, presentes desde la edad pediátrica hasta la vida adulta. Está subdiagnósticado en hombres y puede ser tratado en forma exitosa con glucocorticoides y, en algunos casos, con antiandrógenos. El estudio genético debe ser considerado como una importante herramienta diagnóstica para una adecuada clasificación, especialmente en los probables portadores y para identificar sujetos con alelos con mutaciones graves, considerando el riesgo de tener hijos con la forma clásica de HSC.
Tabla 1. Frecuencia genotípica en la población estudiada
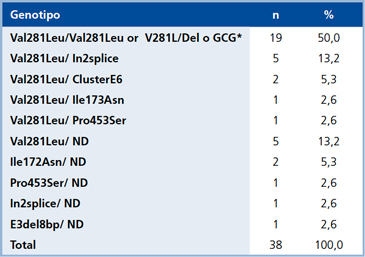
Del: deleción.
GCG: gran conversión génica.
(*) La técnica no distingue entre ambos genotipos.
ND: No Detectado.
Tabla 2. Frecuencia alélica en la población estudiada.
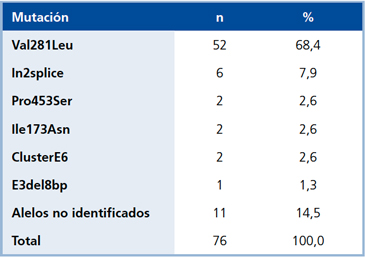
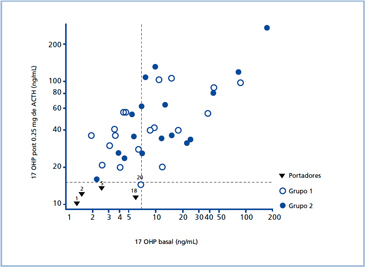 Figura 1. Relación entre 17-OH progesterona basal y posterior al estímulo con 0.25 mg de ACTH.
Figura 1. Relación entre 17-OH progesterona basal y posterior al estímulo con 0.25 mg de ACTH.
La línea segmentada horizontal representa el valor plasmático de 15 ng/mL de 17-OHP y la línea segmentada vertical el valor plasmático de 7 ng/mL de 17-OHP.
Los pacientes identificados con los números 1, 2, 5, 18, 20, se explicitan en la sección de Resultados.
Referencias
- Speiser PW, White PC. 2003 Congenital adrenal hyperplasia. N Engl J Med. 21; 349:776-788.
- New. 2006 Nonclassical 21-Hydroxylase Deficiency. J Clin Endocrinol Metab 91:4205–4214.
- Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. 1985 High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet. 37:650–667.
- Temeck JW, Pang SY, Nelson C, New MI. 1987 Genetic defects of steroidogenesis in premature pubarche. J Clin Endocrinol Metab. 64: 609-617.
- Marynick SP, Chakmakjian ZH, McCaffree DL, Herndon JH Jr. 1983 Androgen excess in cystic acne. N Engl J Med. 28; 308:981-986.
- Dumic M, Ille J, Zunec R, Plavsic V, Francetic I, Skrabic V, et al. 2004 Nonclassic 21-hydroxylase deficiency in Croatia. J Pediatr Endocrinol Metab. 17:157-164.
- Levine LS, Dupont B, Lorenzen F, Pang S, Pollack M, Oberfield S, et al. 1980 Cryptic 21-hydroxylase deficiency in families of patients with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 51:1316-1324.
- Greulich WW, Pyle SI. Radiographics atlas of skeletal development of the hand and wrist. 2nd ed. Standford (CA): Standford Universtity Press, 1959.
- Ferriman D, Gallwey JD. 1961 Clinical assessment of body hair growth in women. J Clin Endocrinol Metab. 21:1440–1448.
- Lahiri D, Nurnberger J. 1991 A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 19:5444.
- Fardella CE, Poggi H, Soto J, Torrealba I, Cattani A, Ugarte F, et al. 2000 Mutations in the CYP21 B gene in a Chilean population with simple virilizing congenital adrenal hyperplasia. J Endocrinol Invest. 23:412-416.
- Fardella CE, Poggi H, Pineda P, Soto J, Torrealba I, Cattani A, et al. 1998 Salt-wasting congenital adrenal hyperplasia: detection of mutations in CYP21B gene in a Chilean population. J Clin Endocrinol Metab. 83: 3357-3360.
- Pineda P, Fardella C, Poggi H, Torrealba I, Cattani A, Soto J, et al. 1997 Molecular diagnosis of salt wasting congenital adrenal hyperplasia, caused by deficit of 21-hydroxylase, in the Chilean population. Rev Med Chil. 125: 987-992.
- Poggi, H.; Soto, J.; Fardella, C.; Romeo, E.; Depix, M.S.; Foradori, A.C. 1999. Molecular Genetic Diagnosis of Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency in 103 Chilean Patients . IFCC Worldlab, Florencia, Italia.
- Pinto G, Tardy V, Trivin C, Thalassinos C, Lortat-Jacob S, Nihoul- Fekete C, et al. 2003 Follow-up of 68 children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: relevance of genotype for management. J Clin Endocrinol Metab 88:2624–2633.
- Azziz R. 1997 Nonclassic adrenal hyperplasia. Curr Ther Endocrinol Metab. 6:175-178.
- Cabrera M, Vogiatzi M, New M 2001 Long term outcome in adult males with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab 86:3070–3080.
- New MI, Gertner JM, Speiser PW, Del Balzo P 1989 Growth and final height in classical and nonclassical 21-hydroxylase deficiency. J Endocrinol Invest 12:91–95.
- David M, Sempe M, Blanc M, Nicolino M, Forest M, Morel Y 1994 Final height in 69 patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Arch Pediatr 1:363–367.
- Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. 1982 Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab 55:817–827.
- Rumsby G, Avey C, Conway G, Honour J 1998 Genotype-phenotype analysis in late onset 21-hydroxylase deficiency in comparison to the classical forms. Clin Endocrinol 48:707–711.
- Feldman S, Billaud L, Thalabard JC, Raux-Demay MC, Mowszowicz I, Kuttenn F, et al. 1992 Fertility in women with late-onset adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 74: 635-639.
- Dewailly D, Vantyghem-Haudiquet M, Sainsard C, Buvat J, Cappoen J, Ardaens K, et al. 1986 Clinical and biological phenotypes in late-onset 21-hydroxylase deficiency. J Clin Endocrinol Metab 63:418–423.
- New MI, Lorenzen F, Lerner AJ, Kohn B, Oberfield SE, Pollack MS, et al. 1983 Genotyping steroid 21-hydroxylase deficiency: hormonal reference data. J Clin Endocrinol Metab. 57: 320-326.
- Speiser PW, New MI 1987 Genotype and hormonal phenotype in nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab, 64:86-91.
- Bachega TA, Billerbeck AE, Marcondes JA, Madureira G, Arnhold IJ, Mendonca BB. 2000 Influence of different genotypes on 17-hydroxyprogesterone levels in patients with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol.52: 601-607.
- White PC, Speiser PW. 2000 Congenital adrenal hyperplasia due 21-hydroxylase deficiency. Endocr Rev 21:245-291.
- Sherman SL, Aston CE, Morton NE, Speiser PW, New MI. 1988 A segregation and linkage study of classical and nonclassical 21-hydroxilase deficiency. Am J Hum Genet 42:830-838.