Cáncer medular de tiroides esporádico de curso agresivo, en un adolescente varón
Rodrigo Bancalari¹, Marcela Molina¹, Pilar Orellana², María Angélica Wietstruck³, Francisco Barriga³, Alejandro Martínez¹, Andreina Cattani¹ y Hernán García B.¹
Medullary thyroid cancer in a 14 years old male.
Case report
¹Departamento Pediatría, Unidad de
Endocrinología, Escuela de Medicina,
Pontificia Universidad Católica de Chile.
²Departamento Radiología, Unidad de
Medicina Nuclear, Escuela de Medicina,
Pontificia Universidad Católica de Chile.
³Departamento Pediatría, Unidad de
Hemato-Oncología, Escuela de Medicina,
Pontificia Universidad Católica de Chile.
Correspondencia a:
Departamento de Endocrinología, Facultad
de Medicina. Pontificia Universidad Católica
de Chile.
Lira 85, 5º piso, Santiago- Chile.
E-mail: rodrigo.bancalari@gmail.com
Recibido: 17 de Noviembre de 2009
Aceptado: 05 de Marzo de 2010
Medullary thyroid cancer can appear sporadically or as part of a multiple endocrine neoplasia type 2A or 2B. In both conditions, it is associated with mutations of proto oncogene RET (rearranged during transfection). We report a 14 years old male presenting with a bone lesion in the skull followed by a hard cervical mass. A CAT scan showed an invasive thyroid nodule with involvement of regional lymph nodes, osteolytic lesions in skull, spine and ribs and liver metastases. Serum calcitonin was markedly elevated (9752 pg/ml, normal below 14 pg/ml). Fine needle biopsy showed a medullary thyroid carcinoma and the patient was subjected to a total thyroidectomy and radical cervical dissection. In the postoperative period the patient required calcium and vitamin D supplementation. Serum calcitonin 15 days after surgery was 11.692 pg/ml. Palliative radiotherapy was indicated for spine pain. A percutaneous gastrostomy was indication for nutritional support. The molecular study did not detect mutations of RET gene between exons 10 and 16.
Key words: Cáncer medular de tiroides, neoplasia endocrina multiple, cáncer medular de tiroides familiar, Proto oncogen RET, calcitonina.
El cáncer medular de tiroides (CMT) es poco frecuente en pediatría y representa entre el 4-8% de todos los cánceres de tiroides1. Este se genera en las células C de la tiroides, las cuales siendo originarias de la cresta neural migran durante la embriogénesis hacia la tiroides. Las células C secretan calcitonina, péptido de 32 aminoácidos que participa en el metabolismo del calcio reduciendo sus niveles plasmáticos, actuando a nivel intestinal, renal y óseo. La hiperplasia multifocal de las células C de la tiroides es la lesión precursora del CMT, la cual puede ser de causa esporádica o presentar una base genética. En 1968 Steiner y cols, acuñan el término neoplasia endocrina múltiple (NEM) para describir la asociación del CMT, hiperparatiroidismo y/o feocromocitoma2.
La NEM tipo 2 es una enfermedad autosómica dominante que presenta dos variantes: NEM 2 A caracterizada por CMT, feocromocitoma e hiperparatiroidismo, y NEM 2 B que presenta CMT y feocromocitoma sin hiperparatiroidismo. Además NEM 2B presenta hábito marfanoide y gangliomas en la mucosa de labio, lengua y ganglioneuromas intestinales3.
En el cáncer medular de tiroides de tipo familiar (CMTF) el CMT se presenta aislado sin otras asociaciones. El CMT esta presente en el 100% de los casos de NEM 2A y 2B siendo la principal causa de muerte en estos pacientes.
Es importante destacar que la aparición del CMT en NEM 2B puede ocurrir antes del año de vida, en NEM 2A alrededor de los 10 años, mientras que en la forma familiar la aparición de CMT en general es más tardía, alrededor de los 18 años, aunque se han descrito casos de aparición más temprana4,5.
Caso clínico
Paciente varón de 14 años de edad, procedente de Antofagasta y con el antecedente de haber presentado a los 8 meses una meningitis bacteriana, quedando con retardo mental leve. Seis meses antes de su hospitalización presenta aumento de volumen duro e indoloro en región frontal y parietal medial del cráneo. Un TAC de cráneo reveló lesiones osteolíticas que se consideraron compatibles con granuloma por presentar I notorio aumento de volúmen de consistencia pétrea, en región cervical izquierda. La ecotomografía tiroidea reveló un nódulo sólido en lóbulo tiroideo izquierdo de 3,2 cm de diámetro mayor con microcalcificaciones, y otro de 1,5 cm de diámetro mayor adyacente a la glándula. Trasladado a nuestro servicio destacó el aumento de volumen pétreo indoloro en región frontal y parietal medial del cráneo, una adenopatía indolora de consistencia aumentada en región cervical izquierda de 2,5 x 1,5 x 3,0 cm. No presentaba neuromas en mucosa oral, y el hábito corporal era normal. Peso 44,5 kg, talla 161 cm (p21) e IMC de 17,2 (p14) (Figura 1). El TAC de cráneo con ventana ósea confirmó la lesión osteolítica a nivel frontal (Figura 2), y el de cuello demostró un nódulo tiroideo izquierdo de aspecto maligno con numerosas adenopatías regionales aisladas, además de lesiones en vértebras cervicales. El TAC de tórax mostró lesiones osteolíticas en vértebras y costillas sin compromiso del parénquima pulmonar y el de abdomen reveló múltiples lesiones focales, hipodensas respecto al resto del parénquima, ubicadas en el segmento posterior del lóbulo hepático derecho, sugerentes de metástasis hepáticas. Cintigrama con Tecnecio 99 y luego un PET-CT con Galio DOTATATE confirma la masa tiroidea con sobreexpresión de receptores de somatostatina asociado a múltiples focos óseos y hepáticos compatibles con metástasis de CMT.
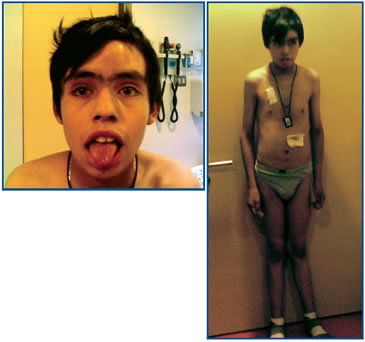 Figura 1. Imágenes facial y corporal. Nótese el aumento
de volumen frontal.
Figura 1. Imágenes facial y corporal. Nótese el aumento
de volumen frontal.
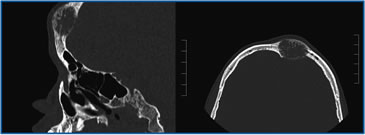 Figura 2. TAC de cráneo con ventana ósea en el que se aprecia una lesión osteolítica en
el hueso frontal.
Figura 2. TAC de cráneo con ventana ósea en el que se aprecia una lesión osteolítica en
el hueso frontal.
Exámenes hormonales: Calcitonina plasmática: 9.725 pg/mL (normal < 13 pg/mL), antígeno carcinoembrionario: 20,3 ng/mL (VN < 5), PTH: 37,3 pg/ mL (15-65 pg/mL) y metanefrinas urinarias: 53 ug/24 h (52-341 ug/24 h), anticuerpos anti tiroglobulina: 237 Ui/ mL y antitiroperoxidasa 605 Ui/mL, T4: 1,87 ng/dL y TSH 2,7 uUI/mL. Otros marcadores tumorales fueron negativos (CA 125 y sub unidad beta de HCG).
La biopsia obtenida por punción con aguja fina fue compatible con CMT. Por ello se realizó tiroidectomía total con resección ganglionar radical de cuello. El estudio histológico de la pieza operatoria confirmó CMT en lóbulo tiroídeo izquierdo (3,3 x 3,0 x 2,5 cm), en ganglios peritirohíodeos (6/18) y en los de grupos II, III y IV izquierdos (1/19).
El paciente evoluciona con hipoparatiroidismo permanente requiriendo a permanencia dosis elevadas de calcitriol y calcio. Presentó parálisis de cuerda vocal con trastornos de la deglución, deterioro nutricional y neumopatías aspirativas que requirieron de gastrostomía percutánea.
A los 2 meses de postoperatorio presenta claudicación e intenso dolor vertebral lumbar, para lo que se administra radioterapia externa en esa zona (2.000 cGy) con lo cual cede el dolor. El estudio molecular de mutaciónes del proto oncogen RET resultó negativo. Se amplificaron 5 fragmentos correspondientes a los exones 10 al 15 del proto oncogen RET localizado en el cromosoma 10 (10q11.2) y luego se realizó la digestión enzimática de los fragmentos amplificados. La secuenciación directa de los exones 10 (codones 609, 611, 618, 620), 11 (codón 634), 13 (codones 768, 790, 791), 14 (codones 804,808), 15 (codones 883,891) no mostró ninguna mutación compatible con NEM tipo 2. Además se amplificó un fragmento de 196 pb que comprende al exón 16 del gen RET, localizado en el brazo largo del cromosoma 10 en la posición 11.2 (10q11.2) y se realizó una secuenciación automática, en la cual no se detectó ninguna mutación en el fragmento analizado compatible con el diagnóstico de MEN tipo 2.
A los 15 días de operado la calcitonina se mantenía elevada (11.692 pg/mL, VN < 13 pg/mL) confirmando persistencia de la enfermedad.
Discusión
Presentamos el caso muy inhabitual de un adolescente de 14 años con CMT el cual al momento del diagnóstico ya presentaba una amplia diseminación de la enfermedad. El paciente no tenía antecedentes familiares de CMT y los valores de metanefrinas urinarias, calcemia y PTH eran normales; tampoco presentaba alteración del tránsito intestinal propia de los ganglioneuromas ni tampoco neuromas visibles al examen físico. El estudio molecular de la mutación del proto oncogen RET descartó un NEM 2 A y NEM 2B; esta última era poco probable en nuestro paciente por no tener hábito marfanoide ni ganglioneuromas; así, se puede establecer el diagnóstico de CMT esporádico. La positividad de los anticuerpos antitiroideos no tiene una explicación clara; es posible que el proceso inflamatorio tumoral que afecta al tiroides, estimule la producción de estos anticuerpos, pero no se puede descartar la coexistencia de una tiroiditis crónica autoinmune con función tiroidea conservada al momento de la consulta.
La importancia del estudio genético radica en el poder dar consejo genético a sus familiares, necesario debido a la gran penetrancia de las mutaciones del RET. La proteína RET es un receptor tirosina kinasa expresado en las células provenientes de la cresta neural que incluye a las células C de la tiroides, las células paratiroideas, células cromoafines de la medula adrenal y células del plexo entérico autonómico. En 1980 fueron descritas las primeras mutaciones en el proto oncogen RET ubicado en el cromosoma 10q11.2 en pacientes portadores de NEM 2 A, NEM 2 B y CMTF6.
Basta la sola mutación activante de un alelo para presentar la neoplasia. En NEM 2 A el codón 634 está mutado en el 87% de los casos, en el NEM 2 B el codón 918 está mutado en el 94% y en el CMTF existe mutación de los codones 618 en el 30%, del 634 en el 26% y del 620 en el 21% de los casos. La lesión inicial es la hiperplasia multifocal de las células C y el tiempo que demora en transformarse en CMT varía según el genotipo, por lo que cobra importancia el estudio genético en los familiares7.
Según el codón afectado se aconseja el momento en el cual realizar la tiroidectomía profiláctica en los familiares del caso índice (Figura 3).
Los pacientes se clasifican en tres grados de riesgo de acuerdo al codón afectado: grado 1 se recomienda tiroidectomía antes de los 10 años de edad; grado 2 antes de los 5 años y grado 3 antes del año de edad. En el caso de un CMT esporádico los familiares tendrían el mismo riesgo que la población general y por lo tanto no hay indicación de tiroidectomía profiláctica8 (Figura 3).
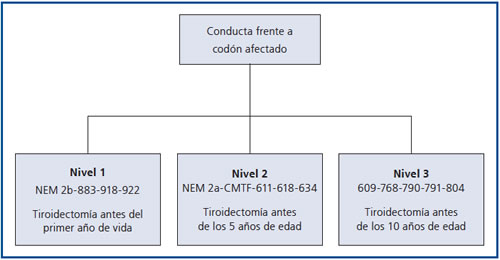
Figura 3. Conducta profiláctica recomendada respecto de los familiares de pacientes con CMT, de acuerdo al codón afectado.
La calcitonina es el principal marcador del CMT y sus niveles presentan una correlación positiva con la masa tumoral, lo que se demostró en nuestro caso que presentaba cifras extraordinariamente elevadas y correspondía a enfermedad con múltiples metástasis.
En el año 2003 Niccoli-Sire et al, observaron que de 35 pacientes portadores de nódulo tiroideo con valores basales elevados de calcitonina (> 4 veces), 34 presentaban CMT y de ellos el 37,1 % con compromiso de ganglionar regional.
Sin embargo, la elevación de la calcitonina no permite diferenciar una hiperplasia multifocal de células C de un CMT, diferenciación que sólo se logra con el estudio histológico9.
Debido a que el CMT es un tumor resistente a la quimioterapia y radioterapia, éstas sólo cumplen una labor paliativa. El tratamiento de elección es la tiroidectomía. Aunque en general es una intervención con pocas complicaciones en equipos quirúrgicos con experiencia, la tasa de hipocalcemia permanente es de 7% y la de daño definitivo del nervio recurrente laríngeo 1 %, lo que puede ser mayor en casos con extensión local10.
Existen actualmente, en etapa de investigación, algunos protocolos de tratamientos para el CMT, enfocados a inhibir la tirosina kinasa o los receptores de los factores de estímulo endotelial vía tirosina kinasa, como el factor de crecimiento endotelial, factor receptor (VEGFR), los cuales tendrían un papel fundamental en la transmisión de señales vía kinasas en mutaciones de proto oncogenes como el RET11.
Nuestro paciente no pudo ingresar a ningún protocolo de investigación con inhibidores de la tirosina kinasa ya que estos, o no incluían pacientes en edad pediátrica o obligaban a permanecer en Estados Unidos, situación no viable en su caso.
Analizando retrospectivamente la conducta terapéutica efectuada en nuestro paciente quien presentaba un retardo mental leve (que dificultó en el post operatorio el entrenamiento para recuperar la deglución normal) y que al momento del diagnostico evidenciaba extenso compromiso cervical y numerosas metástasis a distancia, era discutible obtener beneficios reales con la tiroidectomía total y se pudo haber optado por una cirugía menos extensa, destinada fundamentalmente a prevenir la obstrucción de vía aérea. La extensa cirugía se asoció a un período post-operatorio tórpido y prolongado con constantes microaspiraciones y trastornos de deglución que requirió gastrostomía y dosis altas de calcio y calcitriol por el hipoparatiroidismo instalado.
El CMT es una patología que puede ser muy grave por lo que la precocidad del diagnóstico adquiere gran importancia, así como el estudio genético que permite indicar la tiroidectomía profiláctica único tratamiento con utilidad demostrada, y así salvar la vida de los niños afectados precozmente por esta enfermedad.
Referencias
- Clayman GL, El-Baradie TS. 2003. Medullary thyroid cancer. Otolaryngologic Clinics of North America 36: 91-105.
- Steiner AL, Goodman AD, Powers SR. 1968. Study of a kindred with pheochromocytoma, hyperparathyroidism and Cushing's disease; multiple endocrine neoplasia type 2. Medicine 47: 371-379.
- Skinner MA. 2003. Management of hereditary thyroid cancer in children. Surgical Oncology 12; 101-104.
- Skinner MA, Moley JA, Diley WG, Owzar K, Debenedetti MK, Wells SA Jr. 2005. Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. New England Journal of Medicine 353: 1105-1113.
- Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, et al. 2001. Guidelines for diagnosis and therapy of MEN type 1 and type 2. Journal of Clinical Endocrinology and Metabolism 86: 5658-5671.
- Shaha AR, Cohen T, Ghossein R, Tuttle M. 2006. Late-onset medullary carcinoma of the thyroid: need for genetic testing and prophylactic thyroidectomy in adult family members. Laryngoscope 116: 1704–1707.
- George H, Sakorafas, Helmut Friess1 and George Peros. 2008. The genetic basis of hereditary medullary thyroid cancer: clinical implications for the surgeon, with a particular emphasis on the role of prophylactic thyroidectomy Endocrine-Related Cancer 15: 871-884.
- You YN, Lakhani V, Wells SA. 2007. New directions in the treatment of thyroid cancer. Journal of the American College of Surgeons 205: S45-S48.
- Niccoli-Sire P, Murat A, Rohmer V, Gibelin H, Chabrier G, Conte-Devolx B, et al. 2003. When should thyroidectomy be performed in familial medullary thyroid carcinoma gene carriers with non-cysteine RET mutations? Surgery 134: 1029-1036.
- Rodríguez GJ, Balsalobre MD, Pomares F, Torregrosa NM, Carbonell P, Glower G, et al. 2002. Prophylactic thyroidectomy in MEN 2A syndrome: experience in a single center. Journal of the American College of Surgeons 195: 159-166.
- Steven I, Sherman MD. 2008. Early Clinical Studies of Novel Therapies for Thyroid Cancers Endocrinol Metab Clin N Am 37: 511-524.