Efecto de la activación del Receptor Sensor de Calcio sobre la expresión de genes lipogénicos en células HepG2
Pía Villarroel1,2,3,a y Mariana Cifuentes3,b
Effect of Calcium Sensing Receptor activation on lipogenic gene expression in HepG2 cells
1Escuela de Nutrición y Dietética. Facultad de Ciencias, Universidad Mayor, Santiago, Chile.
2Escuela de Nutrición y Dietética, Facultad de Ciencias de la Salud, Universidad San Sebastián, Santiago, Chile.
3Laboratorio de Nutrición Básica y Epidemiología Genética. Instituto de Nutrición y Tecnología de los Alimentos, Universidad de Chile.
aNutricionista. PhD en Nutrición y Alimentos.
bQuímico Farmacéutico, PhD en Ciencias de la Nutrición. Profesor Titular Universidad de Chile.
Apoyo financiero: Proyecto Sociedad Chilena de
Endocrinología y Diabetes (SOCHED) 2013-04.
Declaración de intereses: Los autores no declaran conflicto
de interés alguno.
Correspondencia:
Pía Villarroel Heise
Escuela de Nutrición y Dietética. Facultad de Ciencias. Universidad Mayor, Campus Huechuraba. Camino la Pirámide 5750, Santiago, Chile.
Teléfono: 23281346
pia.villarroel@mayor.cl - pia.villarroel@inta.uchile.cl
Recibido: 16-05-2017
Aceptado: 19-10-2017
Introduction:The Calcium Sensing Receptor (CaSR) is expressed in human fat cells, and its stimulation may be associated with adipose tissue dysfunction. The multisystemic character of obesity and the search of deepening the scope of the activation of CaSR in this disorder allows us to study the response of this protein in tissues that differ from adipose. Objective: To evaluate the effect of CaSR activation on the expression of lipogenic genes in a model of excess glucose and fatty acids in HepG2 human liver cells. Materials and methods: The effect of the calcimimetic cinacalcet (allosteric agonist of CaSR) on the content of triglycerides (fluorimetry) in a model of glucose supply and on the expression of lipogenic genes (qPCR) in hyperglycemia and hyperlipidemia conditions in the Liver cell line HepG2.
Results: Cinacalcet, glucose (25 mM) and oleic acid (0.6 mM) did not affect cell viability. Activation of CaSR in the presence of glucose failed to increase the intracellular
triglyceride content at 72 hours. Under these conditions, no response
was observed for the factors coding for lipogenic genes (SREBP1c and FAS) at 24
hours of stimulation with cinacalcet in the liver cells. In the case of the over supply
of fatty acids, the HepG2 cells did not show a variation in the gene expression of
the DGAT enzymes after exposure to cinacalcet. Conclusion: Under conditions of
glucose exposure, cinacalcet did not show a response in the triglyceride content,
nor in the expression of genes related to hepatic lipogenesis. Therefore, stimulation
of CaSR would not be associated with hepatic steatosis in HepG2 cells exposed to
glucose.
Key words: Receptor, calcium, lipogenesis, HepG2.
Introducción
En la obesidad, una desregulación en los procesos metabólicos puede resultar en el depósito de grasa ectópica, que se relaciona con la lipogénesis y la esteatosis hepática1. La esteatosis hepática cursa con una alteración en el almacenamiento de triglicéridos en una patología denominada como enfermedad de hígado graso no alcohólica, señalada como uno de los factores claves asociados con insuficiencia metabólica relacionada a la obesidad2.
Se ha descrito la presencia y funcionalidad del Receptor Sensor de Calcio (CaSR) en una variedad de tejidos que difieren de su principal y, originalmente, descrito papel en la homeostasis del calcio3. Nuestro laboratorio ha estudiado la participación del CaSR en las alteraciones en el tejido adiposo que se asocian a las comorbilidades de la obesidad4-7. En el año 2001, Canaff y cols. identificaron por primera vez la expresión del CaSR en el tejido hepático y hepatocitos de cultivo primario de ratas8 y, recientemente, nuestro laboratorio ha descrito la expresión del CaSR en células hepáticas humanas7. Para complementar lo observado en el tejido adiposo, y por su relevancia a nivel sistémico, se estudió la participación del CaSR en la desregulación de la funcionalidad del tejido hepático, sugiriendo que el almacenamiento de triglicéridos en el hígado puede verse alterado ante la activación de esta proteína7. Desde este hallazgo ha surgido la necesidad de estudiar diversos aspectos de la funcionalidad hepática que, potencialmente, estén mediados por la activación del CaSR.
En el contexto del carácter multisistémico de la obesidad y en la búsqueda de profundizar en los alcances de la activación del CaSR en este desorden, este trabajo evaluó si la exposición al calcimimético cinacalcet (agonista alostérico del CaSR) modula la acumulación de triglicéridos en células HepG2 en un modelo de sobreoferta de glucosa, a fin de proponer mecanismos adicionales del CaSR como un factor involucrado en las comorbilidades asociadas a la obesidad.
Materiales y Métodos
Línea celular HepG2
Se utilizó la línea celular HepG2 (American Type Culture Collection, ATCC), derivada de un carcinoma hepatocelular humano y validada como modelo de hepatocitos humanos9. Las células fueron mantenidas en un medio mínimo esencial (MEM), con 10% de suero fetal bovino (SFB) suplementado con antibióticos (1% penicilina/estreptomicina), en una atmósfera controlada (37 °C; 5% CO2).
Reacción en cadena de la polimerasa en tiempo real (qPCR)
El ARN total se obtuvo luego de lisar las células utilizando Trizol® (Invitrogen, Life Technologies INc., Carlsbad, CA, USA). El ARN fue extraído utilizando el Mini Kit PureLink™ RNA (Invitrogen), de acuerdo a las indicaciones del fabricante. La transcripción reversa se realizó usando high Capacity cDNA Reverse Transcription kit (Applied Biosystems, Carlsbad, CA). La expresión de ARNm se evaluó en Step One Real-time PCR System (Applied Biosystems, Foster City, CA, EE. UU.), utilizando SYBR® FAST qPCR Kit (Applied Biosystems). Se utilizaron partidores previamente validados en nuestro laboratorio (Tabla 1). Las condiciones de los ciclos térmicos consistieron en una preincubación de 20 seg a 95 ºC seguido de 40 ciclos de 95 ºC durante 3 segundos y 60 ºC durante 30 seg. Los resultados se normalizaron de acuerdo a la expresión del gen de referencia GAPDH y la expresión se calculó usando el modelo Pfaffl10. Todos los ensayos se realizaron en duplicado.
Target mRNA |
Acceso |
Partidor Forward (5’ ® 3’) |
Partidor Reverse (5’ ® 3’) |
CaSR |
NM_001178065 |
GATGAGACAGATGCCAGTGC |
AAAGAGGGTGAGTGCGATCC |
FAS |
NM_004104.4 |
GGAAGCTGCCAGAGTCGGAGAACT |
TGAGGGTCCATCGTGTGTGCCT |
SREBP1c |
NM_001005291.2 |
ACACAGCAACCAGAAACTCAAG |
AGTGTGTCCTCCACCTCAGTCT |
DGAT1 |
NM_012079.5 |
GAGCTACCCGGACAATCTGA |
AGCTGGGTGAAGAACAGCAT |
DGAT2 |
NM_032564.3 |
GGCAGGCAACTTCCGAATGCCT |
AAAGCCCTTGCGGTTCCGCA |
GAPDH* |
NM_002046 |
GAAGGTGAAGGTCGGAGTCAAC |
CAGAGTTAAAAGCAGCCCTGGT |
*Gen control (Housekeeping).
RT-PCR y electroforesis en gel de agarosa
Luego de extraer el ARN y sintetizar el cDNA, las amplificaciones se realizaron utilizando la enzima Gotaq Master Mix (Promega, Madison, WI, EE. UU.) en el termociclador Biometra® Personal Cycler (Göttingen, Alemania). Las condiciones de los ciclos térmicos consistieron en una preincubación de 2 min a 95 ºC seguido de 40 ciclos a 95 ºC durante 30 segundos, 60 ºC durante 30 segundos y 72 ºC durante 30 segundos. Para terminar con una extensión final a 72 ºC durante 5 min. Como marcador de peso molecular se utilizó DNA Ladder 50 pb (Fermentas, EE. UU.). Se realizó electroforesis en geles de agarosa (1,5% en buffer TBE 1X) entre 40-60 min a 80V, y los amplificados fueron observados bajo luz UV.
Cuantificación del contenido de triglicéridos
Las células fueron tratadas con 0,25% de solución de tripsina e incubadas a 37 ºC por 10 min. Posteriormente, fueron lavadas con PBS e incubadas con Nile Red (1µl/ml) disuelto en PBS por 5-7 min a temperatura ambiente. La fluorescencia fue evaluada utilizando filtros 485 nm de excitación y 572 nm de emisión (fluorímetro Synergy 2 (Biotek®)). Los resultados fueron normalizados por el contenido de proteína medida mediante el método basado en el ácido bicinonínico (Pierce).
Evaluación de la viabilidad celular
Se evaluó viabilidad celular por colorimetría a través del ensayo CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) (Promega, WI, USA) de acuerdo a las condiciones descritas por el fabricante en un espectrofotómetro de escaneo multipocillo ELx808 microplate reader (BioTek Instruments, Inc. VT, USA). Las células HepG2 fueron sembradas a una densidad de 3 x 103 células/cm2. Después de 24 h de incubación se realizó un cambio de medio y se inició el tratamiento con los distintos agentes por 72 h. Posteriormente, cada pozo fue tratado con 20 μl del reactivo 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) y se incubó por 3 h a 37 ºC. El colorante de formazán, producido por las células metabólicamente activas, se cuantificó a través de la medición de absorbancia a una longitud de onda de 490 (restándole la lectura a 630 nm para descartar el background de acuerdo con las recomendaciones del fabricante).
Resultados
Evaluación del efecto de la activación del CaSR sobre el contenido de triglicéridos en la línea celular HepG2 en un modelo de sobreoferta de glucosa
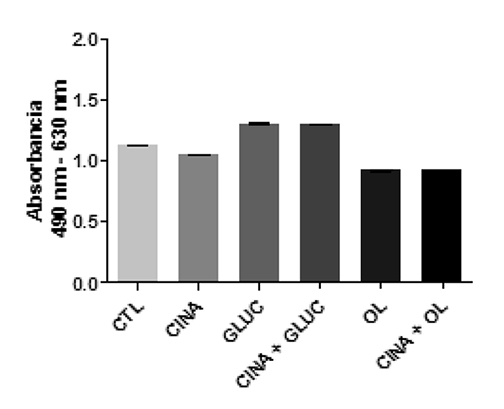
Cuando la capacidad del tejido adiposo de almacenar grasa se ve sobrepasada por un exceso de oferta de nutrientes, puede producirse un depósito de lípidos en el hígado. Este hecho da lugar a una alteración deletérea del metabolismo en este órgano, con consecuencias clínicas importantes. Utilizando un modelo de sobreoferta de nutrientes para observar el manejo de triglicéridos en células HepG2, expusimos las células a concentraciones consideradas altas (25 mM) de glucosa11 o (0,6 mM) ácido oleico12. Para descartar muerte celular dado el riesgo de toxicidad ante estas altas concentraciones, se determinó la viabilidad de estas células frente a los estímulos (Figura 1).
Una vez establecido que los tratamientos usados en las concentraciones indicadas no afectan la viabilidad celular, se evaluó el efecto de cinacalcet, glucosa y ácido oleico sobre la acumulación de triglicéridos (Figura 2).
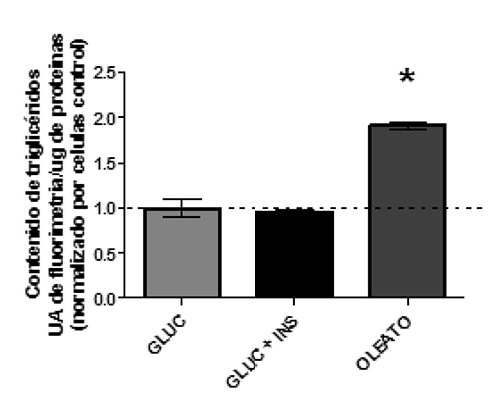
La Figura 2 muestra que el tratamiento con ácido oleico aumenta significativamente el contenido de triglicéridos comparado con el cultivo control, mientras que la concentración de lípidos intracelulares no respondió frente al tratamiento con glucosa (con o sin insulina). Posteriormente, se estimuló las células con cinacalcet para evaluar el efecto de la activación del CaSR en las condiciones de sobreoferta glicémica (Figura 3).
La Figura 3 muestra que la activación del CaSR no afecta el contenido de triglicéridos en presencia de glucosa. No obstante, en presencia de ácido oleico la estimulación del CaSR por 72 h aumenta significativamente la concentración de lípidos intracelulares7. Esperando explicar estas diferencias se evaluó el efecto de la estimulación del CaSR sobre la respuesta génica de factores lipogénicos como DGAT1/2 (asociados a la sobreoferta de lípidos) y de SREBP1c y FAS (asociados a la sobreoferta de glucosa) en los modelos antes mencionados. Los genes propuestos son de particular interés por representar distintas etapas de la lipogénesis hepática, hecho que podría ser vinculado a los efectos sistémicos de la inflamación y disfunción adiposa asociadas a la obesidad.
Efecto de la activación del CaSR sobre la expresión génica de factores lipogénicos en células HepG2
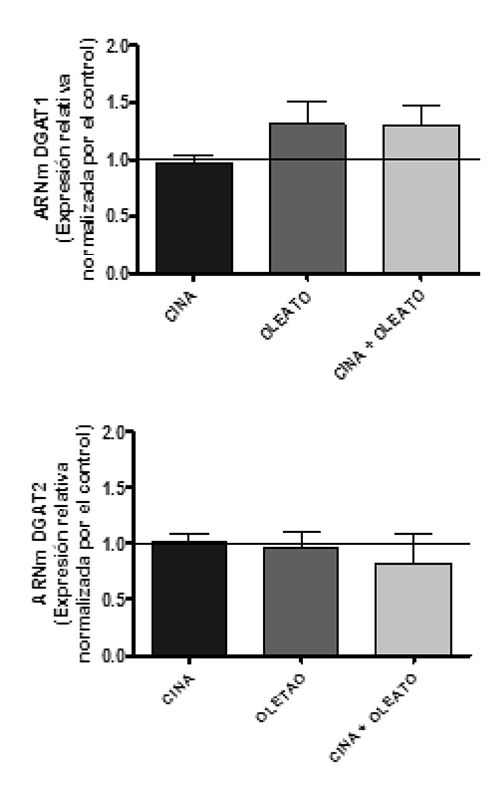
Previamente, se reportó un aumento en el contenido de triglicéridos para células HepG2 expuestas a ácido oleico7, por lo tanto, es interesante estudiar la respuesta de los genes lipogénicos DGAT1/2 en estas condiciones, dado que ellos participan en etapa final de la síntesis de triglicéridos (Figura 4).
Para aquellas células expuestas a ácido oleico, observamos en la Figura 4 que este estímulo no afecta la expresión del ARNm de DGAT1/2. No se observó un efecto de cinacalcet sobre la respuesta génica de DGAT1/2.
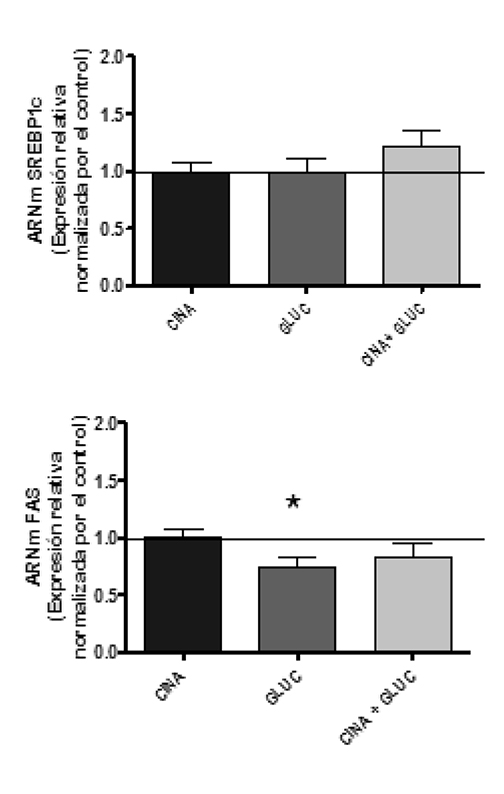
Por otro lado, se estudió la respuesta de SREBP1c y FAS en un contexto de hiperglicemia. Estos factores participan en la lipogénesis de novo, respaldando el uso del exceso crónico de glucosa como sustrato para la síntesis de triglicéridos (Figura 5).
Como muestra la Figura 5, la exposición de células HepG2 a glucosa y/o cinacalcet no muestra una respuesta de SREBP1c. En esta última condición es interesante destacar que FAS disminuye significativamente su expresión cuando las células hepáticas se exponen a glucosa comparadas con el cultivo control. No se observó una respuesta en la expresión de SREBP1c o FAS ante la estimulación del CaSR.
Discusión
Los resultados obtenidos permiten proponer un modelo en el que, identificado el CaSR en células hepáticas humanas y en consistencia con lo reportado previamente7, la proteína CaSR no participaría en el desarrollo de la esteatosis hepática en condiciones de sobreoferta de glucosa. Además, hemos podido establecer que, al comparar la expresión génica de factores lipogénicos, entre el modelo de sobreoferta de glucosa y de sobreoferta de lípidos, la estimulación del CaSR no influye en la respuesta de los genes propuestos. Por tanto, sugerimos que el estímulo del CaSR, en un contexto hiperglicémico, no contribuiría (de acuerdo al mecanismo estudiado) al desarrollo de comorbilidades asociadas a la obesidad.
Nuestras observaciones están exponiendo algunas manifestaciones de la célula hepática en respuesta a la activación del CaSR en dos contextos fisiopatológicos diferentes (sobreoferta de lípidos y sobreoferta de glucosa). La relación existente entre la sobreoferta crónica de glucosa (hiperglicemia) y el consecuente aumento de la lipogénesis de novo13, nos habría permitido interpretar este modelo como una manifestación de disfuncionalidad hepática. Al observar la presencia de lípidos intracelulares en células hepáticas humanas HepG2, evidenciamos que la activación del CaSR no aumenta el contenido de triglicéridos en presencia de glucosa, respuesta significativamente incrementada en células expuestas a ácido oleico7.
Entre las principales funciones metabólicas del hígado se encuentran la síntesis, almacenamiento, secreción y catabolismo de ácidos grasos y triglicéridos, que pueden ser evaluados, entre otras, a través de la expresión de enzimas y factores de transcripción relevantes en las distintas vías, tales como la proteína 1 ligante al elemento regulador de esteroles (SREBP-1c), la sintasa de los ácidos grasos (FAS) y las diaciglicérido aciltranferasas 1 y 2 (DGAT1/2). En el caso de la sobreoferta de ácidos grasos se estudiaron 2 enzimas (DGAT1 y DGAT2), que a pesar de catalizar la misma reacción (síntesis de triglicéridos a partir de la acil-CoA y diacilglicerol)14, se sugiere que DGAT2 posee mayor afinidad por ciertos sustratos que DGAT115. Se ha establecido que DGAT1 juega un papel más importante en la esterificación de ácidos grasos exógenos, mientras que DGAT2 es principalmente responsable de la incorporación de ácidos grasos de síntesis endógena a los triglicéridos16. A pesar de la falta de respuesta sobre la expresión de DGATs por efecto de cinacalcet, estudios anteriores7 han podido establecer un posible mecanismo mediado por la inflamación para sugerir la etiopatogénesis de la esteatosis hepática frente a una sobre carga lipídica.
Sobre la base de que la hiperglicemia incrementaría una respuesta de genes asociados a la lipogénesis de novo (SREBP-1c y FAS)13, y que la estimulación del CaSR podría asociarse a este fenotipo negativo en el hígado7, podríamos especular que las condiciones experimentales utilizadas deberían respaldar un aumento en la expresión de los genes estudiados. No obstante, cinacalcet no muestra el efecto esperado, la activación del CaSR no aumenta SREBP1c y/o FAS en células hepáticas en presencia de glucosa. SREBP-1c es un factor de transcripción que puede ser estimulado por glucosa17, el gen más importante en la regulación de la lipogénesis de novo en el hígado y responsable de la activación de genes implicados en la biosíntesis de ácidos grasos, incluyendo FAS18. Adicionalmente, la glucosa también es capaz de estimular otro factor de trascripción como ChREBP (proteína ligadora de elementos de respuesta a carbohidratos), que induce no solo enzimas glucogénicas, sino que también enzimas lipogénicas como FAS19. Por esto, la sintasa de ácidos grasos debiera haber respondido al estímulo de glucosa en las células hepáticas de manera dependiente o independiente a SREBP1c. Sin embargo, se debe tener presente la compleja respuesta fisiológica asociada a eventos de hiperglicemia, con lo que tampoco se puede descartar que al evaluar una otra ventana de tiempo pudiera evidenciarse una respuesta diferente de los genes estudiados. Asimismo, la falta de respuesta observada puede fundar la existencia de otros mecanismos generados por la hiperglicemia (como resistencia a la insulina) que estén bloqueando el desarrollo de lipogénesis hepática.
En el hígado, la sobreoferta lipídica se relaciona con un aumento en el contenido de triglicéridos hepáticos7, mientras que la sobreoferta de glucosa no mostró esta tendencia. Esto podría sugerir que la hiperlipidemia exacerbaría el fenotipo negativo de la obesidad en mayor magnitud que la hiperglicemia. En otras palabras, si el CaSR participa como mediador en la generación de estatosis hepática, podríamos discutir o pensar que un sujeto dislipidémico debería verse más propenso/dañado a una esteatosis hepática que un sujeto diabético.
Nuestros resultados apoyan la relevancia del estudio de una conversación cruzada adiposo-hepático, pues el tejido adiposo provee de ácidos grasos que el hígado capta13. Estudios anteriores de nuestro laboratorio han podido mostrar que la estimulación del CaSR en el tejido adiposo sería responsable de una menor lipogénesis5, involucrando posiblemente una mayor cantidad de ácidos grasos circulantes, susceptibles de ser captados por el hígado. Cuando se estimula el CaSR en células hepáticas expuestas a ácido oleico, se promueve un mayor depósito de triglicéridos en este tejido7, permitiendo postular al CaSR como mediador entre la disminución de la lipogénesis adiposa y el aumento de la lipogénesis hepática. En este contexto patológico podemos especular un probable efecto deletéreo del CaSR sobre el hígado asociado a una exposición a ácidos grasos que no fue posible observar en condiciones de exposición a glucosa.
Agradecimientos
Los autores agradecen el apoyo de la Sociedad Chilena de Endocrinología y Diabetes (SOCHED) proyecto 2013-04 y del Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) 1150651
Referencias bibliográficas
- Ginsberg H, Zhang Y, Hernández-Ono A. 2006. Metabolic syndrome: Focus on dyslipidemia. Obesity 14: 41-9.
- Diraison F, Dusserre E, Vidal H, Sothier M, Beylot M. 2002. Increased hepatic lipogenesis but decreased expression of lipogenic gene in adipose tissue in human obesity. Am J Physiol Endocrinol Metab 282, E46eE51.
- Brown E, Gamba G, Riccardi D, Lombardi M, Butters R, K ifor O. et al. 1993. Cloning and characterisation of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 366: 575-580.
- Cifuentes M, Fuentes C, Tobar N, Acevedo I, Villalobos E, Hugo E, et al. 2012. Calcium sensing receptor activation elevates proinflammatory factor expression in human adipose cells and adipose tissue. Molecular and cellular endocrinology 361: 24-30.
- Villarroel P, Reyes M, Fuentes C, Segovia M, Tobar N, Villalobos E, et al. 2013. Adipogenic effect of calcium sensing receptor activation. Molecular and cellular biochemistry 384: 139-145.
- Rocha G, Villalobos E, Fuentes C, Villarroel P, Reyes M, Díaz X, et al. 2015. Preadipocyte proliferation is elevated by calcium sensing receptor activation. Molecular and cellular endocrinology 412: 251-256.
- Villarroel P, Mattar P, D’Espessailles A, Arrese M, Arreguin A, Fuentes C, et al. 2016. Calcium sensing receptor effects in adipocytes and liver cells: Implications for an adipose-hepatic crosstalk. Arch Biochem Biophys 607: 47-54.
- Canaff L, Petit J, Kisiel M, Watson P, Gascon-Barré M, Hendy G. 2001. Extracellular calcium-sensing receptor is expressed in rat hepatocytes coupling to intracellular calcium mobilization and stimulation of bile flow. Biol Chem 276: 4070-9.
- Bokhari M, Carnachan R, Cameron N, Przyborski S. 2007. Culture of HepG2 liver cells on three dimensional polystyrene scaffolds enhances cell structure and function during toxicological challenge. J Anat 211: 567-76.
- Pfaffl M. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45.
- Khodabukus A, Baar K. 2015. Glucose concentration and streptomycin alter in vitro muscle function and metabolism. J Cell Physiol 230: 1226-34.
- Kwan H, Fong W, Yang Z, Yu Z, Hsiao W. 2013. Inhibition of DNA-dependent protein kinase reduced palmitate and oleate-induced lipid accumulation in HepG2 cells. Eur J Nutr 52: 1621-30.
- Geisler C, Renquist B. 2017. Hepatic lipid accumulation: cause and consequence of dysregulated glucoregulatory hormones. J Endocrinol. pii: JOE-16-0513. doi: 10.1530/JOE-16-0513 [Epub ahead of print].
- Liu Y, Millar J, Cromley D, Graham M, Crooke R, Billheimer J, et al. 2008. Knockdown of acyl-CoA: diacylglycerol acyltransferase 2 with antisense oligonucleotide reduces VLDL
- Yen C, Stone S, Koliwad S, Harris C, Farese RV Jr. 2008. Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res 49: 2283-301.
- Qi J, Lang W, Geisler J, Wang P, Petrounia I, Mai S, et al. 2012. The use of stable isotope-labeled glycerol and oleic acid to differentiate the hepatic functions of DGAT1 and -2. J Lipid Res 53: 1106-16.
- Palou M, Priego T, Sánchez J, Villegas E, Rodríguez A, Palou A, et al. 2008. Sequential changes in the expression of genes involved in lipid metabolism in adipose tissue and liver in response to fasting. Pflugers Arch 456: 825-36.
- Iizuka K, Bruick R, Liang G, Horton J, Uyeda K. 2004. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci USA 101: 7281-6.