Consideraciones en el manejo del adulto con Hiperplasia Suprarrenal congénita clásica por déficit de 21-Hidroxilasa
Amanda Ladrón de Guevara H.1,2*, Mariano Smith G.1,3
Management considerations for the adult with congenital adrenal hyperplasia due 21-hydroxylase deficiency
- Laboratorio de Endocrinología y Metabolismo, Facultad de Medicina Occidente, Universidad de Chile. Santiago, Chile.
- Unidad de Endocrinología Servicio de Medicina, Hospital San Juan de Dios. Santiago, Chile.
- Alumno de Medicina cuarto año. Universidad de Chile. Santiago, Chile.
*Correspondencia:
Amanda Ladrón de Guevara Hernández
E-mail: a.ldeguevara@yahoo.com
Las Palmeras 299, interior
Quinta
Normal.
Recibido: 14-09-2020
Aceptado: 29-12-2020
Resumen: La Hiperplasia Suprarrenal Congénita (HSRC) corresponde a un grupo de defectos genéticos en la síntesis de cortisol. El 95% de ellas son debidas al déficit de 21-hidroxilasa por lo que nos referiremos solo a esta deficiencia. La hiperplasia suprarrenal congénita clásica (HSRC-C) debuta en recién nacidos o lactantes con insuficiencia suprarrenal primaria, diferentes grados de hiperandrogenismo clínico en mujeres y puede coexistir con hipotensión, hiperkalemia e hiponatremia si hay un déficit clínico de aldosterona. El objetivo de este artículo es actualizar el conocimiento y enfoques sugeridos para el manejo de la HSRC-C desde el inicio de sus controles en la etapa adulta. El diagnóstico diferencial en retrospectiva de la HSRC-C y la no clásica (HSRC-NC) a veces resulta difícil ya que esta enfermedad es un espectro fenotípico continuo. La insuficiencia suprarrenal y la dependencia a terapia corticoidal son los eventos principales para diferenciar estas dos patologías que tienen enfoques terapéuticos diferentes. El tratamiento de la HSRC-C en adultos abarca 2 objetivos primarios: la adecuada sustitución de la falla suprarrenal y el control de hiperandrogenismo mediante el uso de corticoides en sus dosis mínimas efectivas. En la mujer existen terapias complementarias para el control del hiperandrogenismo como anticonceptivos y otras que se encuentran en diferentes fases de investigación. Esto permite disminuir las dosis de corticoides en algunos casos. Es importante a la vez abordar tres objetivos secundarios: controlar el riesgo cardiometabólico propio de la enfermedad, evitar el sobre tratamiento corticoidal y manejar la infertilidad. La correcta monitorización del tratamiento en adultos tomando en cuenta los objetivos descritos permite una mejor calidad de vida en estos pacientes. Finalmente el consejo genético debe realizarse en todos los pacientes con HSRC que deseen fertilidad y en sus parejas. El estudio requiere de secuenciación del gen CYP21A2 y debe realizarse en un laboratorio de experiencia.
Palabras clave: Aldosterona; Hiperandrogenismo; Hiperplasia adrenal congénita; Insuficiencia Adrenal; 21-Hidroxilasa.
Abstract: Congenital Adrenal Hyperplasia (CAH) are a group of genetic defects characterized by impaired cortisol synthesis. 95% of them are due to 21-hydroxylase deficiency. We will discuss only this enzyme’s deficiency. Classic congenital adrenal hyperplasia (CAH-C) debuts in newborns or infants with primary adrenal insufficiency, some degree of clinical hyperandrogenism in newborn females, and can coexist with hypotension, hyperkalemia, and hyponatremia if there is a clinical aldosterone deficiency. The objective of this article is to update the knowledge and suggested approaches for the management of CAH-C from the beginning of its controls in the adult stage. The retrospective differential diagnosis of CAH-C and non-classical (CAH-NC) is sometimes difficult because this disease is a continuous phenotypic spectrum. Adrenal insufficiency and dependence on corticosteroid therapy are the main events to differentiate these two pathologies that have different therapeutic approaches. In adults, the treatment of CAH-C must include 2 primary objectives: adequate the replacement of adrenal failure and control of hyperandrogenism, through the use of corticosteroids in their minimum effective doses. In women there are complementary therapies for the control of hyperandrogenism, such as contraceptives and others that are in different phases of research. This makes it possible to reduce the doses of corticosteroids in some cases. It is important at the same time to address three secondary objectives: control the cardiometabolic risk of the disease secondary to corticosteroid treatment, avoid corticosteroid overtreatment and manage infertility. The correct monitoring of treatment in adults and taking in to account the objectives described, allows a better quality of life in these patients. Finally, genetic counseling must be carried out in all patients planning for children, with any type of CAH and in their partners. The study requires sequencing of the CYP21A2 gene and must be performed in a certified laboratory.
Keywords: Adrenal insufficiency; Aldosterone; Congenital adrenal hyperplasia; Hyperandrogenism; Steroid 21-Hydroxylase.
La Hiperplasia Suprarrenal Congénita (HSRC) corresponde un grupo de defectos genéticos, autosómicos recesivos que alteran síntesis de cortisol. El 95% de ellas corresponden al déficit de 21-hidroxilasa (21OH). En esta revisión nos referiremos a la HSRC por déficit de 21OH ya que es la enzima más comúnmente afectada. Esta puede presentarse en sus formas Clásicas (HSRC-C) las cuales requieren tratamiento con glucocorticoides y/o mineralocorticoides de por vida; o en su forma más leve, No Clásica (HSRCNC), la que no requiere sustitución.
La HSRC-C por este déficit se divide en perdedora de sal ya que presenta un alteración pronunciada en la producción de aldosterona y en no perdedora de sal o virilizante simple, en donde existe un déficit de aldosterona que no se manifiesta clínicamente por lo tanto, ambas tienen una alteración en la función de esta hormona en mayor o menor grado1.
Del punto de vista fisiopatológico, la HSRC-C debuta en recién nacidos o lactantes con una insuficiencia suprarrenal primaria que presenta algún grado de hiperandrogenismo desde la gestación, secundario al incremento de ACTH que estimula la vía indemne de producción androgénica suprarrenal. Por lo tanto, en la etapa adulta, tendremos un paciente con talla baja por estímulo excesivo de andrógenos que cierran precozmente el cartílago de crecimiento, hiperpigmentación de la piel por el aumento de POMC (secundario a la pérdida de retroalimentación negativa que proporciona el cortisol) e hiperandrogenismo clínico (genitales ambiguos en sexo femenino, hirsutismo y en casos extremos virilización). Asociado a esto puede existir hipotensión, hiperkalemia e hiponatremia si existe un déficit clínico de aldosterona2.
Estos pacientes han mejorado notablemente sus expectativas de vida desde que se inició la corticoterapia en los años 50-60 y cuando se comenzó la pesquisa en recién nacidos, en los años 903.
El objetivo de este artículo es la actualización del conocimiento y enfoques sugeridos para el manejo de estos pacientes en la adultez.
Diagnóstico en el adulto
El diagnóstico en la adultez presenta 2 desafíos: La atención en el servicio de urgencia y la primera consulta ambulatoria.
En la primera situación el paciente se encuentra habitualmente con un evento agudo y cierto grado de compromiso de conciencia, por lo que generalmente no aporta una buena historia. Sin embargo, tenemos la clínica descrita previamente para reconocerlos y no perder tiempo en iniciar el reemplazo corticoidal.
La segunda situación es aún más desafiante ya que la mayoría de los pacientes aporta datos incompletos de su historia clínica. Por otra parte, no es fácil diferenciar en retrospectiva entre la HSRC-C no perdedora de sal de la HSRC-NC. Esta última es una forma menos grave de la misma enfermedad. Si en la clásica existe un 0 a 2% de funcionalidad enzimática, la no clásica presenta un 20 a 50% de actividad de la 21OH. Como esta enfermedad es un espectro fenotípico continuo hay un punto intermedio en el cual es difícil diferenciar ambas patologías1,4.
La HSRC-NC frecuentemente debuta con pubarquia prematura. El fenómeno fisiopatológico principal es el hiperandrogenismo de inicio más tardío. Por lo anterior, tampoco existirán genitales ambiguos. Pueden presentar talla baja, pero esta es menos pronunciada5 y es extremadamente infrecuente la insuficiencia suprarrenal2.
En cuanto al estudio, las guías sugieren medir 17-hidroxiprogesterona (17OHP) basal, tanto para el diagnóstico como para el seguimiento. Para el diagnóstico, esta debe ser tomada en Fase Folicular del ciclo (entre el 3er a 8vo día de regla), en ausencia de anticoncepción hormonal. En caso de duda puede agregarse un test de estímulo con ACTH para 17OHP (Figura 1)2.
En la práctica clínica esto es mucho más confundente, debido al amplio espectro fenotípico de esta enfermedad. Los resultados de este test deben ser interpretados según los signos y síntomas de cada sujeto. Podemos tener pacientes con HSRC-NC con 17OHPG basal >10 ng/ml con talla baja e hiperandrogenismo evidente, ausencia de sexo ambiguo y sin historia de falla suprarrenal, así como podemos ver otros con enfermedad clásica sin hirsutismo, con talla baja, sexo ambiguo al nacer y falla suprarrenal marcada.
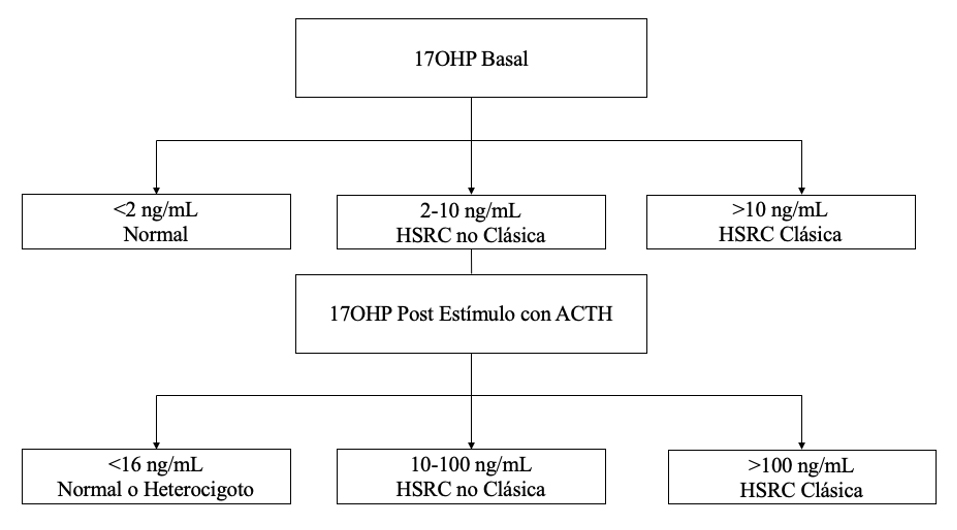 Figura 1: Algoritmo Diagnóstico Basado en 17OHP.
Figura 1: Algoritmo Diagnóstico Basado en 17OHP.Objetivos del tratamiento
Existen dos objetivos primarios:
- Reemplazar la falla suprarrenal
- Reducir la secreción de CRH-ACTH para controlar hiperandrogenismo.
En la etapa adulta también hay que cumplir con tres objetivos secundarios:
- Abordar el riesgo cardiometabólico
- Evitar sobre tratamiento corticoidal
- Manejo de la infertilidad.
Tratamiento corticoidal
La meta es lograr concentraciones lo más fisiológicas posibles de cortisol, cumpliendo a su vez con los dos objetivos primarios.
No hay estudios controlados a largo plazo para indicar el tratamiento corticoidal ideal en adultos con HSRC-C dada la amplia variabilidad de respuesta al tratamiento. Por otro lado en el ser humano existe una sensibilidad idiosincrática al uso de corticoides2.
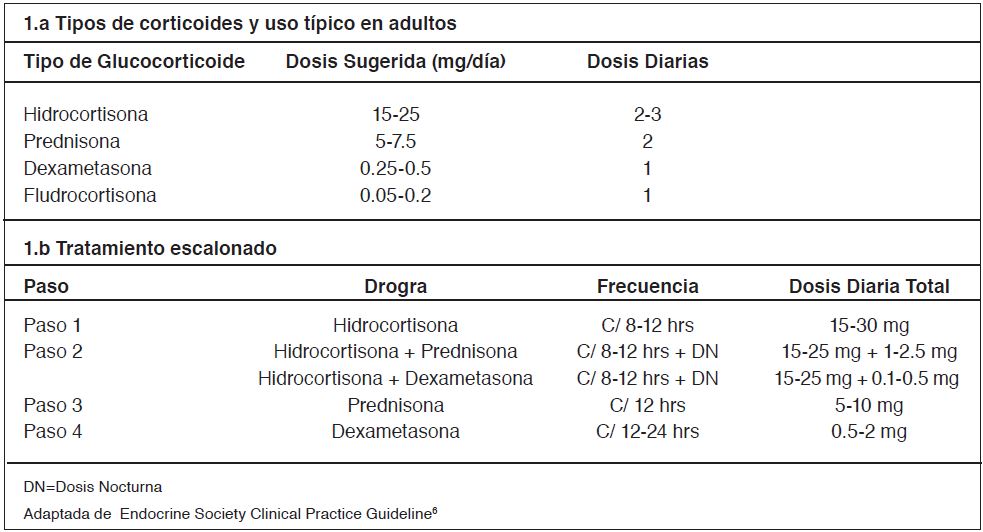
En la tabla 1 (1.a) se muestra el uso terapéutico de corticoides en HSRC-C.
Existen pacientes que presentan un predominio de falla suprarrenal muy importante, con gran dependencia al tratamiento de reemplazo, pero con un hiperandrogenismo no muy marcado. Hay otros en los que predomina el hiperandrogenismo por sobre la falla suprarrenal. Existen pacientes, sobre todo varones, que pueden permanecer varios días sin tratamiento de reemplazo corticoidal, pero frente a un evento agudo tendrán una crisis. Lo mismo ocurre con la necesidad de mineralocorticoides6.
Hidrocortisona
Es el corticoide más fisiológico y más utilizado en esta enfermedad. Además, es el que menos produce síndrome de Cushing.
La dosis de 20 mg diarios tiene una actividad Requiere de 2 a 3 dosis, por lo que frecuentemente los pacientes olvidan alguna. Debido a su vida media corta, muchas veces no logra frenar el pick de ACTH, que se genera entre las 3 y 4 AM.
La hidrocortisona en sus dosis habituales presenta peaks suprafisiológicos y nadires infrafisiológicos, no pudiendo reproducir el ritmo circadiano normal7.
Tratamiento corticoidal: Sistema escalonado
El tratamiento escalonado es el que se ha utilizado históricamente en esta enfermedad. El enfoque es similar al que se tiene frente a un paciente diabético o hipertenso (Tabla 1(1.b)).
Se debe partir con un fármaco simple y seguro que permita suplir la falla suprarrenal (paso 1). Luego se van agregando terapias progresivamente más potentes, equilibrando siempre el riesgo versus el beneficio (paso 2 ,3 ó 4). No se debe controlar el hiperandrogenismo a expensas de un sobretratamiento corticoidal6.
Hay que intentar imitar el ciclo circadiano de cortisol, ya que mejora la sensación de bienestar, reduce la ACTH y esteroides suprarrenales. La dosis de ritmo diurno invertido, que busca frenar 17OHP, se debe evitar, ya que aumenta el riesgo de enfermedades metabólicas8.
En el paso 2 por ejemplo, se puede agregar dosis bajas de prednisona nocturna, utilizando la dosis mínima efectiva para controlar el hiperandrogenismo. Si no existe un buen control de este, se puede agregar un fármaco con mayor efecto en la frenación de la ACTH. Es importante mencionar que la dexametasona es el corticoide más potente y debe restringirse solo para circunstancias especiales como restos adrenales o infertilidad rebelde a tratamiento (paso 4), siempre considerando que en el paso 2 en la mayoría es bastante efectivo6,8.
Considerar también que gran porcentaje de pacientes de “difícil manejo”, son en realidad falta de adherencia a tratamiento.
Tabla 1. Corticoterapia en HSRC-C.
Mineralocorticoides
El reemplazo del déficit de aldosterona se logra a través de un régimen con sal, asociado a fludrocortisona (0,1 a 0,2 mg/ día vía oral). Con estas 2 acciones se logra reestablecer las concentraciones de sodio y potasio, se estabiliza la presión arterial (evitando la hipotensión ortostática) y se regula la actividad de renina plasmática (PRA)9.
La necesidad de mineralocorticoides, por lo general, decrece con la edad y se mantiene relativamente estable durante la adultez. La subdosificación de estas medidas puede aumentar la Angiotensina II, lo que estimula los primeros pasos de la vía esteroidogénica, incrementando la síntesis de andrógenos suprarrenales. También puede aumentar la ADH y la ACTH, generando hiponatremia. Además, el reemplazo óptimo de mineralocorticoides pudiera reducir la dosis de glucocorticoides diarios6. Hay que recordar que la hidrocortisona es el único corticoide con actividad aldosterónica en las dosis terapéuticas utilizadas en esta enfermedad.
Manejo no corticoidal de Hiperandrogenismo en la mujer
Anticoncepción Hormonal e Hiperandrogenismo
Esta se usa para evitar embarazo y como manejo del hiperandrogenismo. Hay que tomar en cuenta los riesgos habituales para una mujer sana o con riesgo cardiometabólico según el caso.
La terapia combinada resulta muy útil, ya que aumenta la SHBG, protege el hueso y logra un buen control de ciclos.
El uso de Valerato de Estradiol es ideal por su buen perfil metabólico, baja incidencia de trombosis y protección ósea10.
En cuanto a los progestágenos, debe evitarse el uso de antialdosterónicos. Las guías son claras evitar el uso de espironolactona, pero no con respecto a su derivado, drospirenona6. Ambos fármacos inhiben la 17-hidroxilasa suprarrenal. Se cita un estudio en 22 mujeres sanas que usaron drospirenona en anticoncepción combinada, sin presentar bajas de presión11. Esta publicación parece ser insuficiente para sugerir drospirenona en una paciente con HSRC que puede tener algún grado de déficit de aldosterona, clínicamente aparente o no. Sin embargo, si la paciente ya está usando un anticonceptivo combinado con este progestágeno y es asintomática puede mantenerlo.
Dienogest un progestágeno derivado de gonanos, resulta ser una buena alternativa para combinar con estrógenoterapia por su buen efecto antiandrogénico ya que es capaz de elevar la SHBG e inhibir a la 5areductasa, siendo una molécula neutra en cuanto a riesgo metabólico y de menor riesgo trombótico que las progesteronas de 3ra y 4ta generación10.
Flutamida
Es un fármaco antiandrogénico no esteroidal, bloqueador de receptor de testosterona y dehidrotestosterona. Además aumenta la degradación hepática de testosterona. Se recomienda que sea prescrito sólo por especialistas con experiencia debido a su hepatoxicidad12.
Nuevas terapias
Existen 2 grandes líneas:
- Imitar las dosis circadianas de cortisol mediante hidrocortisona de liberación lenta.
- Bloquear el hiperandrogenismo por vías alternativas, para disminuir las dosis terapéuticas de corticoides.
1. Imitar dosis circadianas de cortisol
Chronocort
Es una hidrocortisona de liberación retardada, en donde la dosis nocturna es la responsable del incremento de cortisol durante la madrugada, mientras que la dosis de 10 mg matinal mantiene los niveles diurnos. Este esquema imita satisfactoriamente al ciclo circadiano13.
El estudio de Mallappa (Figura 2), muestra que los niveles de ACTH plasmáticos fueron muy similares a los de la hidrocortisona en 3 dosis, incluso logra frenar mejor esta hormona. También controló mejor la 17OHP y androstenediona con respecto al tratamiento clásico. La fase 3 de este estudio debe terminarse en 2021, por lo que podría aprobarse13.
Bomba de hidrocortisona
Durante el 2016 se publicó un artículo que utilizó una bomba de insulina para la infusión continua de cortisol. El objetivo de este estudio fue evaluar la seguridad y la eficacia de la bomba en pacientes con HSRC-C de difícil manejo14.
La bomba se aproximó a la secreción fisiológica de cortisol. Además, comparado con glucocorticoides orales, redujo los niveles de ACTH, 17OHP, androstenediona y progesterona. Desde el punto de vista metabólico, los resultados fueron positivos. Se produjo aumento de la osteocalcina, c-telopéptido y mejoró la masa magra. Otro hallazgo fue que 1 de las 3 mujeres amenorreicas reanudó la menstruación y 1 hombre tuvo reducción del TART, lo que es destacable ya que las concentraciones totales de cortisol fueron menores que en el uso de hidrocortisona en 3 tomas. En el 2018 se publicó el seguimiento en 6 de los 8 pacientes. Hasta el momento ha demostrado ser seguro, bien tolerado y con estabilidad en los resultados a los 18 meses15.
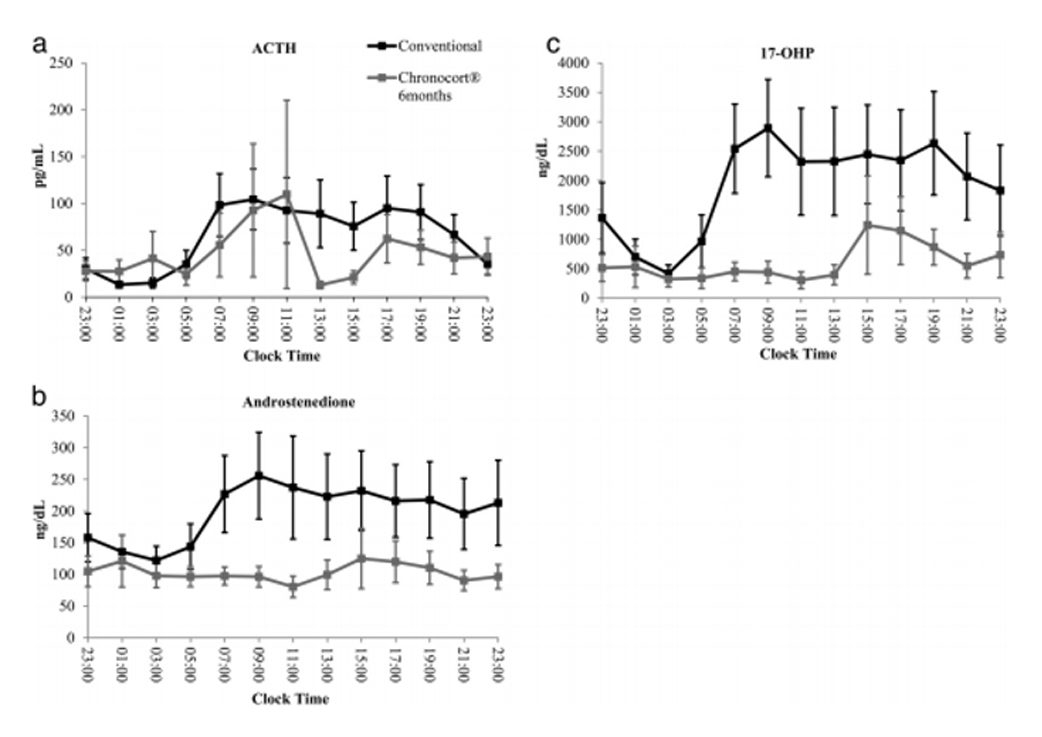 Figura 2: Comparación del perfil hormonal en terapia de hidrocortisona (ver dosis) en 3 tomas versus Chronocort (20 mg nocturnos + 10 mg diurnos).
Figura 2: Comparación del perfil hormonal en terapia de hidrocortisona (ver dosis) en 3 tomas versus Chronocort (20 mg nocturnos + 10 mg diurnos).2. Terapias no corticoidales
- Abiraterona: es un inhibidor de CYP17A1, utilizado en el cáncer de próstata, que bloquea la síntesis de andrógenos. En el caso de HSRC-C en pacientes de sexo femenino, hay un estudios en fase 1, en los cuales 250 mg han logrado normalizar la androstenediona3,16,17.
- Antagonista Selectivo del Receptor de CRH de Tipo 1 NBI-77860: hay un estudio en fase 1 en 8 pacientes, en donde 600 mg a las 22.00 hrs reduce la ACTH en 40% y la 17OHP en 30%18.
- Mitotane: este fármaco podría restaurar la fertilidad en pacientes varones con TART. El Mitotane tiene un efecto adrenalítico, además de inhibir la esteroidogénesis. Se publicó el caso de un paciente de 29 años con 2 años de infertilidad al momento del estudio. Después de 8 meses de tratamiento, mejoraron los niveles de gonadotropinas, la Inhibina B y el recuento de espermatozoides. Hubo una reducción del tamaño de los TART, lo que se atribuye a la acción adrenalítica del Mitotane. El paciente que presentaba azoospermia inicialmente, logró embarazo por fertilización in vitro con recién nacido de término, heterocigota para el gen de 21OH19.
Objetivos secundarios
1. Control del riesgo cardiometabólico
Se ha observado que las enfermedades cardiovasculares son la primera causa de muerte después de los 50 años en estos pacientes.
La prevalencia de enfermedades cardiometabólicas en la HSRC-C presenta un OR de 3,9 comparado con sujetos normales, independiente del IMC20.
Estudios previos han evidenciado que estos pacientes desarrollan precozmente resistencia a la insulina (RI) y riesgo cardiovascular, independiente de la dosis acumulada de glucocorticoides teniendo mayor prevalencia de HTA, FA, TVP, obesidad, DM2 y SAHOS6,20. El objetivo es la prevención y manejo oportuno de estas condiciones que se presentan en la HSRC-C en la etapa adulta.
El último metaanálisis publicado al respecto incluyó 14 estudios, de los cuales solamente 7 de ellos eran en adultos (300 niños /adolescentes y 137 adultos). Se observó un incremento significativo en la PA en niños con HSRC21.
Se ha evidenciado tanto en humanos como en modelos animales, que la exposición crónica y excesiva a andrógenos altera la actividad y disminuye la producción de óxido nítrico, aumentando la HTA. También incrementa la RI en estos sujetos22.
Basado en este metaanálisis, no se encontró evidencia directa de cardiopatía, morbilidad o mortalidad metabólica asociadas con HSRC. Estos autores concluyen que en la HSRC “podría” existir mayor riesgo cardiometabólico en comparación con controles. Sin embargo, el resultado pudo haberse modificado por la gran cantidad de sujetos jóvenes que incluían y a que solo un estudio evaluaba la mortalidad. Así, esta guías recomiendan un control cardiometabólico, según las pautas convencionales para sujetos sanos6.
Según este análisis, evaluar anualmente la morbilidad metabólica de estos pacientes, y tratar a quien corresponda.
2. Exceso de terapia corticoidal
En los consensos se enfatiza en que el exceso de corticoides produce osteoporosis. Falhamar, en su estudio de registro, describe una osteopenia en 40% y osteoporosis en 7% de los pacientes, con mayor incidencia de fractura, en comparación a controles sanos. Otros autores no encuentran mayor incidencia de fracturas, pero sí mayor incidencia de osteoporosis6,23.
La gravedad de la osteoporosis está relacionada con la mayor dosis de corticoides. Chakhtoura calculó la dosis acumulada de tratamiento en estos pacientes, con la que ya existe un riesgo fractura, medido por T-Score en densitomería ósea. Observó que el riesgo de osteoporosis aumenta significativamente con más de los 100 mg/m2 de corticoides acumulado, lo que representa alrededor 15 mg de hidrocortisona diarios durante 20 años de uso24. Por lo tanto, cuando ingrese un paciente al primer control de adulto, debería solicitarse una densitometría ósea basal. Los pacientes tienen que suplementarse calcio, vitamina D y mantener ejercicio constante6. Por otra parte, es sabido que el uso de corticoides va a producir obesidad central, síndrome metabólico y síndrome de Cushing. Esto siempre se empeora con la dosis inversa. Además, estos pacientes tienen mayor porcentaje de DM2 y desarrollan diabetes gestacional en un 20%6,25.
Manejo de la infertilidad
Fertilidad femenina
El 25% de las mujeres con HSRC-C y 10% de las más severas buscan embarazo debido a que deben sortear una serie de dificultades. Algunas de estas pacientes han tenido una clitoroplastía durante la infancia y secundario a esto una denervación del clítoris perdiendo así su única fuente de orgasmo26.
La mayoría de las pacientes presentan estrechez vaginal en la adultez. El tratamiento de dilatación es doloroso y lento. Esto sumado a talla baja e hiperandrogenismo genera una desmotivación en la búsqueda de pareja.
Por otro lado, las mujeres presentan altos niveles de progestágenos plasmáticos de origen suprarrenal, lo que genera alteraciones en la mucosa cervical y endometrio. Si la paciente desea embarazo, se debe lograr un progesterona en Fase Folicular < 0,6 ng/mL para evitar la acción anticonceptiva progestágena que tiene la suprarrenal sobre el útero27.
Finalmente hay que tratar la oligoanovulación de estas pacientes. El hiperandrogenismo suprarrenal va a generar una alteración de la sensibilidad del hipotálamo a la progesterona, modificando la pulsatilidad de GnRH aumentando la LH, la que estimula a la célula de la teca ovárica produciendo mayor cantidad de hormonas masculinas. Esta también es estimulada por el efecto LH símil dado por la hiperinsulinemia que presentan estas pacientes. Por otro lado, el exceso de andrógenos produce un aumento de la actividad 5a reductasa en el ovario, generando andrógenos más potentes. Todo esto va a generar un Síndrome de Ovario Poliquístico Secundario, que debe tratarse para lograr la fertilidad26.
Fertilidad masculina
La infertilidad masculina también es multifactorial, pero se puede resumir en 2 factores: alteraciones anatómicas y niveles altos de andrógenos suprarrenales.
Las alteraciones anatómicas están dadas por los TARTs, que por efecto masa van a impedir el flujo sanguíneo, dificultan la salida de semen y generan fibrosis testicular. Estas masas de origen adrenal son de crecimiento progresivo y pueden aparecer tardíamente. Se requiere de terapia corticoidal efectiva para frenar la ACTH, evitando así el estímulo de esta sobre los tejidos ectópicos26.
Por otro lado, los altos niveles de andrógenos suprarrenales van a frenar la secreción de LH y FSH impidiendo el estímulo de estas sobre la producción de testosterona de origen testicular y sobre la espermatogénesis respectivamente, provocando atrofia testicular, alteración espermática y, en fases finales fibrosis del teste. Por último, el aumento de la FSH y la disminución de Inhibina-B representan un signo ominoso desde el punto de vista de fertilidad16,26.
Monitorización del tratamiento en adultos
Tratamiento corticoidal
La medición de 17OHP plasmática es el estándar para el diagnóstico y control. Es una hormona sensible en niños, sin embargo, su correlación con andrógenos en adultos es variable. Es necesario solicitar androstenediona en conjunto para tener una mejor guía. Por otro lado, la supresión o normalización de esta hormona indica exceso de tratamiento. Se deben mantener niveles de 17OHP y androstenediona levemente elevados. Los rangos óptimos no han sido definidos y deben correlacionarse con la clínica6. En pacientes de sexo femenino sin uso de anticonceptivos, pueden tolerarse niveles de 17OHP matinales elevados si estas presentan ciclos menstruales regulares, hiperandrogenismo controlado y ausencia de falla suprarrenal.
En conclusión, es importante ajustar la dosis según clínica y objetivos a cumplir.
Para logran un mejor control, se están estudiando nuevos metabolitos más estables, como el 21-deoxicortiso,11-oxiesteroide en plasma6 así como 17OHP, 11-hidroxiandrostenediona y 11-Ketostestosterona salivales28.
Reemplazo de mineralocorticoides
Es indispensable medir la PA y objetivar la hipotensión ortostática. Controlar al paciente con electrolitos plasmáticos, función renal evitando la hipo e hiperkalemia y la hiponatremia.
Es deseable controlar la relación aldosterona/PRA matinal anual, evitando el exceso de renina9. Aspectos clínicos y de laboratorio relevantes Se sugiere medir parámetros antropométricos en cada control (peso, IMC, índice cintura cadera). Controlar perfil metabólico una vez al año y tratar las comorbilidades que se presenten.
El estudio con imágenes como TAC o resonancia no son sugeridos salvo que se presente una clínica o bioquímica atípica. El 4% de estos pacientes presenta un nódulo suprarrenal que habitualmente resulta ser un mielolipoma6.
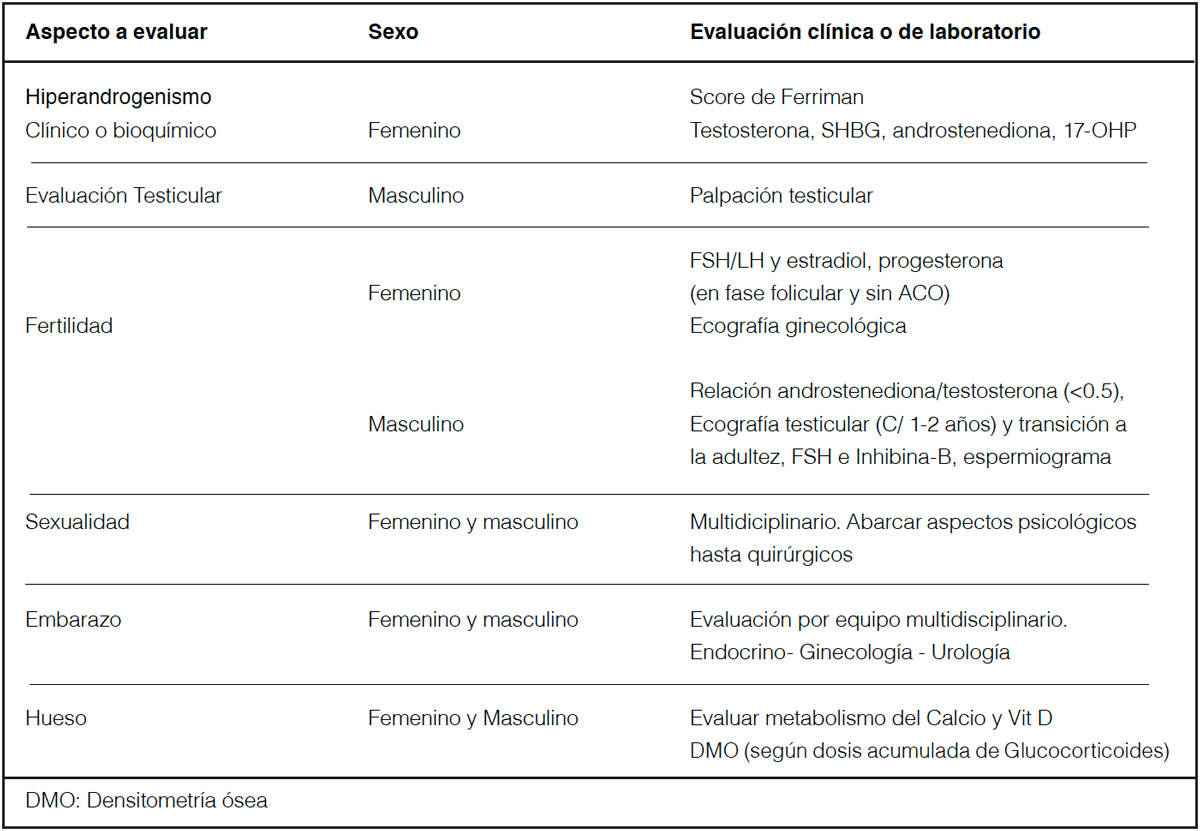
En la tabla 2 se describe la monitorización clínica y de laboratorio con respecto al hiperandrogenismo, a la salud reproductiva y del hueso.
En varones siempre realizar palpación testicular. Si estos son grandes y dolorosos pueden indicar presencia de TART. También pueden encontrarse muy pequeños y fibróticos, lo que refleja mal pronóstico de fertilidad6.
Es necesario medir la relación androstenediona/testosterona, Si esta relación es > 2 refleja mayor producción de andrógenos suprarrenales por sobre la testicular.
La ecografía testicular se solicita cada 1 ó 2 años, ya que TART son progresivos en el tiempo y pueden ir apareciendo a través de los años.
Si se busca fertilidad, medir FSH e Inhibina-B junto con espermiograma6.
Respecto a la sexualidad, esta es sumamente compleja,
e incluye aspectos psicológicos y quirúrgicos. Hay mayor
incidencia en disforia de género en este grupo de pacientes6.
Tabla 2. Monitorización clínica y de Laboratorio de la Salud Reproductiva y ósea en pacientes con HSRC-C.
Consejo genético
La HSRC es una enfermedad autosómica recesiva. Hay un 25% de probabilidades de que los hermanos del caso índice tengan HSRC y 50% de probabilidad de que sean portadores asintomáticos.
En cuanto a la HSRC-NC, aproximadamente dos tercios de los pacientes son heterocigotos compuestos, portando un alelo que causa HRSC-C y uno que causa la no clásica. La mutación más leve determinará el fenotipo. De esta manera, un padre con HSRC-NC tiene un riesgo de 1/240 de tener un hijo con HSRC-C. Esto sube en la práctica clínica a 2,5%, por las parejas portadoras.
Por este motivo, deben estudiarse todos los pacientes que deseen fertilidad y sus parejas con cualquier tipo de HSRC. El estudio requiere de secuenciación del gen CYP21A2 y hacerlo en un laboratorio de experiencia29.
Agradecimientos: Queremos agradecer la valiosa ayuda de la Dra. Teresa Sir Petermann en la redacción de esta revisión y por su inagotable espíritu docente.
Referencias
- Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Lateonset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1982; 55(5): 817-827.
- Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. A Summary of the Endocrine Society Clinical Practice Guidelines on Congenital Adrenal Hyperplasia due to Steroid 21-Hydroxylase Deficiency. Int J Pediatr Endocrinol. 2010; 2010: 494173.
- Auchus RJ, Witchel SF, Leight KR, Aisenberg J, Azziz R, Bachega TA, et al. Guidelines for the Development of Comprehensive Care Centers for Congenital Adrenal Hyperplasia: Guidance from the CARES Foundation Initiative. Int J Pediatr Endocrinol. 2010; 2010: 275213.
- . Witchel SF. Nonclassic congenital adrenal hyperplasia. Curr Opin Endocrinol Diabetes Obes. 2012; 19(3): 151-158.
- Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibanez L, et al. 21-Hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder: a multicenter study. Am J Obstet Gynecol. 2000; 183(6): 1468- 1474.
- Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018; 103(11): 4043-488.
- Simon N, Castinetti F, Ouliac F, Lesavre N, Brue T, Oliver C. Pharmacokinetic evidence for suboptimal treatment of adrenal insufficiency with currently available hydrocortisone tablets. Clin Pharmacokinet. 2010; 49(7): 455-463.
- Riepe F. Summary of the workshop “Management of CAH: the relevance of steroids in plasma, saliva and urine”. Pediatr Endocrinol Rev. 2011; 9(Suppl 1): 541-543.
- Nimkarn S, Lin-Su K, Berglind N, Wilson RC, New MI. Aldosterone-to-renin ratio as a marker for disease severity in 21-hydroxylase deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2007; 92(1): 137-142.
- Fruzzetti F, Cagnacci A. Venous thrombosis and hormonal contraception: what’s new with estradiol-based hormonal contraceptives? Open Access J Contracept. 2018; 9: 75-79.
- de Nadai MN, Nobre F, Ferriani RA, Vieira CS. Effects of two contraceptives containing drospirenone on blood pressure in normotensive women: a randomized-controlled trial. Blood Press Monit. 2015; 20(6): 310-315.
- Martin KA, Anderson RR, Chang RJ, Ehrmann DA, Lobo RA, Murad MH, et al. Evaluation and Treatment of Hirsutism in Premenopausal Women: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018; 103(4): 1233-1257.
- Mallappa A, Sinaii N, Kumar P, Whitaker MJ, Daley LA, Digweed D, et al. A phase 2 study of Chronocort, a modified-release formulation of hydrocortisone, in the treatment of adults with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2015; 100(3): 1137-1145.
- Nella AA, Mallappa A, Perritt AF, Gounden V, Kumar P, Sinaii N, et al. A Phase 2 Study of Continuous Subcutaneous Hydrocortisone Infusion in Adults With Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab. 2016; 101(12): 4690-4698.
- Mallappa A, Nella AA, Sinaii N, Rao H, Gounden V, Perritt AF, et al. Longterm use of continuous subcutaneous hydrocortisone infusion therapy in patients with congenital adrenal hyperplasia. Clin Endocrinol (Oxf). 2018; 89(4): 399-407.
- Bachelot A, Grouthier V, Courtillot C, Dulon J, Touraine P. Management of endocrine disease: Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: update on the management of adult patients and prenatal treatment. Eur J Endocrinol. 2017; 176(4): R167-R81.
- Turcu AF, Auchus RJ. Novel treatment strategies in congenital adrenal hyperplasia. Curr Opin Endocrinol Diabetes Obes. 2016; 23(3): 225-232.
- Turcu AF, Spencer-Segal JL, Farber RH, Luo R, Grigoriadis DE, Ramm CA, et al. Single-Dose Study of a Corticotropin-Releasing Factor Receptor-1 Antagonist in Women With 21-Hydroxylase Deficiency. J Clin Endocrinol Metab. 2016; 101(3): 1174-1180.
- Bry-Gauillard H, Cartes A, Young J. Mitotane for 21-hydroxylase deficiency in an infertile man. N Engl J Med. 2014; 371(21): 2042-2044.
- Falhammar H, Frisen L, Hirschberg AL, Norrby C, Almqvist C, Nordenskjold A, et al. Increased Cardiovascular and Metabolic Morbidity in Patients With 21-Hydroxylase Deficiency: A Swedish Population-Based National Cohort Study. J Clin Endocrinol Metab. 2015; 100(9): 3520-3528.
- Tamhane S, Rodriguez-Gutierrez R, Iqbal AM, Prokop LJ, Bancos I, Speiser PW, et al. Cardiovascular and Metabolic Outcomes in Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab. 2018; 103(11): 4097-4103.
- Muthusamy K, Elamin MB, Smushkin G, Murad MH, Lampropulos JF, Elamin KB, et al. Clinical review: Adult height in patients with congenital adrenal hyperplasia: a systematic review and metaanalysis. J Clin Endocrinol Metab. 2010; 95(9): 4161-4172.
- Falhammar H, Filipsson H, Holmdahl G, Janson PO, Nordenskjold A, Hagenfeldt K, et al. Fractures and bone mineral density in adult women with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007; 92(12): 4643-4649.
- Chakhtoura Z, Bachelot A, Samara-Boustani D, Ruiz JC, Donadille B, Dulon J, et al. Impact of total cumulative glucocorticoid dose on bone mineral density in patients with 21-hydroxylase deficiency. Eur J Endocrinol. 2008; 158(6): 879-887.
- Dallman MF, Akana SF, Bhatnagar S, Bell ME, Strack AM. Bottomed out: metabolic significance of the circadian trough in glucocorticoid concentrations. Int J Obes Relat Metab Disord. 2000; 24 Suppl 2: S40-S46.
- Auchus RJ. Management considerations for the adult with congenital adrenal hyperplasia. Mol Cell Endocrinol. 2015; 408: 190-197.
- Casteras A, De Silva P, Rumsby G, Conway GS. Reassessing fecundity in women with classical congenital adrenal hyperplasia (CAH): normal pregnancy rate but reduced fertility rate. Clin Endocrinol (Oxf). 2009; 70(6): 833-837.
- Bacila I, Adaway J, Hawley J, Mahdi S, Krone R, Patel L, et al. Measurement of Salivary Adrenal-Specific Androgens as Biomarkers of Therapy Control in 21-Hydroxylase Deficiency. J Clin Endocrinol Metab. 2019; 104(12): 6417-6429.
- Hannah-Shmouni F, Chen W, Merke DP. Genetics of Congenital Adrenal Hyperplasia. Endocrinol Metab Clin North Am. 2017; 46(2): 435-458.