Rol del hierro en el desarrollo de diabetes mellitus tipo 2
Mónica Andrews G. y Miguel Arredondo O.
Role of iron in type 2 diabetes mellitus
Laboratorio de Micronutrientes, Instituto de Nutrición y Tecnología de los Alimentos (INTA), Universidad de Chile.
El Líbano 5524, Macul,
Santiago Chile.
Correspondencia a:
Mónica Andrews G.
El Líbano 5524, Macul
Santiago, Chile.
E-mail: monica.andrews@gmail.com
Recibido: 23 de abril de 2012
Aceptado: 18 de mayo de 2012
Iron is an essential nutrient but its excessive levels are potentially dangerous. High body iron levels are associated with increased level of oxidative stress. Epidemiological studies have shown a relationship between increased levels of ferritin or iron stores with the risk of developing type-2 diabetes. Iron-induced oxidative stress would be the most plausible mechanism causing tissue damage in the liver, muscle, adipose tissue and pancreatic β cells. Iron causes increased glucose output by the liver, increased lipolysis, insulin resistance in adipose tissue and β cell dysfunction, resulting in a decreased ability to secrete insulin.
Key words: Diabetes mellitus type 2, iron, ferritin, oxidative stress.
Introducción
El hierro (Fe) es un nutriente esencial en el ser humano y tanto los estados de deficiencia o exceso de Fe resultan en enfermedad. El Fe es un cofactor de varias enzimas envueltas en reacciones oxido reducción debido a su habilidad de existir en dos formas iónicas: en su forma ferrosa (Fe+2) y/o férrica (Fe+3)1. Esta característica hace que el Fe sea potencialmente peligroso en exceso, ya que puede inducir la producción de especies reactivas al oxígeno (ROS), que aumentan la toxicidad celular y el daño oxidativo de componentes celulares tales como proteínas, lípidos y ADN2.
Recientemente, en sujetos portadores de hemocromatosis hereditaria se ha demostrado que existe una relación entre el aumento de las reservas de Fe, en forma de ferritina y el desarrollo de diabetes mellitus tipo 2 (DM2)3. Esta enfermedad se asocia por un aumento de las reservas de Fe en tejido hepático y en otros tejidos como corazón, páncreas y cerebro. Por otro lado, estudios longitudinales han mostrado que sujetos con DM2 tienen mayores niveles circulantes de ferritina que individuos no diabéticos4, correlacionándose, además, estos niveles con los niveles de insulina y glucosa circulantes5,6, hipertensión5, dislipidemias7 y obesidad8. El objetivo de esta revisión es mostrar la evidencia existente sobre el rol del Fe en el desarrollo y/o complicaciones de la resistencia a la insulina y de la diabetes mellitus tipo 2.
Metabolismo del hierro
Como ya se mencionó, el hierro es un micronutriente esencial y sus reservas corporales provienen principalmente de la dieta. El Fe de la dieta se encuentra en dos formas: el Fe hem que se encuentra en productos de origen animal (carnes rojas principalmente) y el Fe no hem derivado de vegetales y legumbres. A pesar que el Fe hem no es predominante en la dieta, es el que mejor se absorbe en el tracto gastrointestinal (25% de absorción del Fe hem vs 5-15% Fe no hem)9.
La captación de Fe por parte del enterocito es altamente regulada, ya que no existe un sistema fisiológico de excreción del metal. Es así como, a mayor reservas de Fe en forma de ferritina la captación del mineral disminuye10. El Fe no hem es captado en el enterocito por el Transportador de Metales Divalentes 1 (DMT1). Como la mayor parte del Fe inorgánico de la dieta esta en la forma Fe3+, éste debe ser reducido antes de ser captado por el DMT1, lo que es llevado a cabo por una reductasa del ribete estriado denominada Duodenal Cytochrome C Reductase B (DcytB). (Figura 1)11.
La absorción del Fe hem es más eficiente que la del Fe no hem. El grupo hem proveniente de Hb y mioglobina es liberado por la pepsina y HCl en el estómago y por las enzimas pancreáticas del lumen intestinal, quedando estabilizado por péptidos productos de la degradación de la globina junto con otros componentes de la dieta. Un posible transportador del Fe hem es la Heme Carrier Protein 1 (HCP1), esta proteína es altamente expresada en el intestino, principalmente en duodeno y también en hígado y riñón, a pesar de la evidencia que relaciona a HCP1 con el transporte apical del Fe hem ha sido también demostrado que esta proteína transporta más eficientemente folatos que Fe hem12. Una vez que el hem alcanza el citoplasma, es degradado por la enzima microsomal hem oxigenasa (HO), produciendo CO, biliverdina y Fe+2 libre. Existen tres isoformas de HO que catalizan la oxidación de hem utilizando NADPH y NADPH-citocromo P-450 reductasa. Por cada molécula de hem oxidada se utilizan tres moléculas de O2 y tres de NADPH. La biliverdina producida es rápidamente reducida a bilirrubina por la biliverdina IXα reductasa (Figura 1)13. El Fe captado, indistintamente de su origen y dependiendo de las demandas corporales el Fe es almacenado en forma de ferritina en la célula intestinal o es transportado por la membrana basolateral del enterocito a través del transportador de salida Ferroportina (Fpn), siendo así entregado para su transporte por la circulación a la transferrina en la forma de Fe3+ 14.
Existen dos tipos de Fe circulante: el Fe unido a transferrina y el no unido a transferrina. Este último, es importante ya que posee un alto potencial redox, pudiendo participar en la reacción de Fenton con la generación de ROS14. Para entregar el hierro a la célula, la holo-transferrina se une a su receptor (Receptor para Transferrina: RTf), complejo que es incorporado a la célula por un proceso de endocitosis10.
El Fe es utilizado principalmente en la médula ósea para la eritropoyesis y en una menor medida por otros los tejidos para proliferación, diferenciación y como parte de numerosas enzimas. La vida media de los glóbulos rojos es de aproximadamente 120 días, transcurrido este tiempo, los glóbulos rojos senescentes son eliminados de la circulación por el sistema retículo endotelial y el Fe así liberado, es reciclado y utilizado para la formación de nuevas células1. El Fe es almacenado en forma de ferritina y hemosiderina principalmente en el hígado, pero también en una menor proporción en la médula ósea y el bazo. Estas proteínas de almacenamiento de Fe constituyen un tercio del total del Fe corporal. La ferritina sérica es utilizada comúnmente para la determinación del estatus de Fe de un individuo. Los niveles de esta proteína se correlacionan positivamente con el Fe no unido a transferrina15 y negativamente con el RTf16. Sin embargo, la especificidad de la ferritina como un marcador del estado nutricional de Fe hoy en día es discutida, ya que por ser una proteína de fase aguda sus niveles se ven alterados en presencia de inflamación17.
Entonces, el balance de Fe es regulado por la captación intestinal, el reciclaje de Fe y la movilización hepática. Recientemente, se ha demostrado que el metabolismo de Fe es regulado estrechamente por la hormona denominada Hepcidina (Hpc), que es sintetizada principalmente en el hígado y en menor grado en las células intestinales, adipocito y células β pancreáticas. La función de la Hpc es regular negativamente la vía de entrada del Fe a la circulación18, es decir, inhibir el paso de hierro desde los enterocitos y macrófagos hacia la circulación, a través del bloqueo de la actividad del transportador de salida Ferroportina (Figura 1).
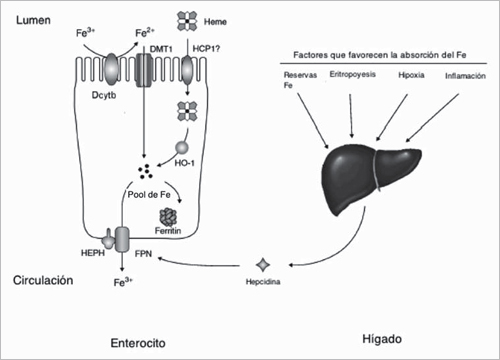
Figura 1. Captación de Hierro (modificado de Anderson et al., 2009). HO-1: Hem Oxigenasa; HEPH: Hefestina; FPN: Ferroportina; HCP1: Hem Carrier Protein 1; Dcytb: Duodenal Cytochrome C Reductase B; DMT1: Transportador de Metales Divalentes 1.
Estrés oxidativo y hierro
Los mecanismos reguladores de la absorción de Fe tienen como objetivo prevenir un exceso del mineral en la circulación disminuyendo así la posibilidad de formar especies reactivas al oxígeno (ROS). Los efectos tóxicos del Fe libre se relacionan con su capacidad de catalizar la generación de radicales libres vía la reacción de Haber-Weiss y la reacción de Fentón14:
Reacción de Haber-Weiss: Fe3+ + •O2 ![]() Fe2+ + O2
Fe2+ + O2
Reacción de Fenton: Fe2+ + H2O2 ![]() Fe3+ + OH- + •OH
Fe3+ + OH- + •OH
La reacción de Fenton tiene una importante participación en condiciones de sobre-carga de Fe, debido a la formación del radical hidroxilo, el cual tiene una vida media corta, (1 nanosegundo), sin embargo, éste reacciona en los sitios cercanos a su formación. La producción este radical cerca del DNA desencadena daño en el material genético19.
El Fe amplifica el daño oxidativo, los órganos con mayor actividad mitocondrial son los más afectados por los ROS, entre ellos, las células del sistema nervioso central, hepatocitos, cardiomiocitos y células β pancreáticas20. En células pancreáticas de ratón con sobrecarga de Fe, se ha observado un aumento del estrés oxidativo, aumento de la apoptosis y disminución de la capacidad de secretar insulina. La célula β es especialmente sensible al daño oxidativo debido a que tiene una disminuida dotación de enzimas antioxidantes tales como la superóxido dismutasa (SOD), la catalasa y de glutatión21.
Relación hierro y diabetes mellitus tipo 2
Estudios epidemiológicos han mostrado una asociación entre sobrecarga de Fe y resistencia a la insulina periférica. En un estudio realizado en 1.013 hombres Finlandeses, se encontró que comparado con el menor quintil de los niveles de ferritina, aquellos que se encontraban en el mayor quintil tenían mayores niveles de insulina y de glucosa circulantes22. Por otro lado, en un estudio prospectivo realizado en 1.277 adultos franceses, se observó que los niveles ferritina fueron un factor predictivo independiente del incremento de los niveles de insulina basal al final de 3 años; por cada 1 ng/ml de incremento de ferritina había un aum ento de insulina de 0,05 (± 0,02) μU/ml23.
La obesidad y el síndrome metabólico son factores de riesgo para el desarrollo de DM2 y en ambas se han observado niveles aumentados de Fe circulantes. En la Encuesta Nacional de Salud y Nutrición de Estados Unidos (NHANES) realizada entre los años 1984 y 1994, se reportó una asociación positiva entre los niveles séricos de ferritina y el IMC de hombres y mujeres. Además, se encontró una correlación positiva entre la circunferencia abdominal y otros índices de distribución de grasa corporal con los niveles de ferritina independiente de la edad y del IMC en hombres entre 20 y 49 años24.
Recientemente, se ha propuesto como un componente del síndrome metabólico, la presencia de elevados niveles de ferritina25, esto debido a lo observado en el estudio NHANES, donde el aumento de depósitos de Fe se asoció a la presencia de síndrome metabólico. Además, los resultados del Estudio Epidemiológico del Síndrome de Resistencia a la Insulina (DESIR) mostraron que altos niveles de ferritina y transferrina se asociaban con una aumentada prevalencia de síndrome metabólico y además eran predictivos de la incidencia de éste síndrome en un seguimiento de 6 años17.
El modelo más estudiado en la relación Fe y diabetes es el de hemocromatosis hereditaria, desorden genético resultante de la mutación del gen HFE que conduce a la no expresión de la proteína HFE en la superficie celular. La proteína HFE es requerida para la estimulación de la síntesis hepática de Hepcidina, la que regula el ingreso de Fe al sistema a través de la internalización y degradación lisosomal de la Ferroportina en enterocitos y macrófagos18. La alteración de la degradación de Ferroportina en la hemocromatosis desencadena una entrada descontrolada de Fe a la circulación, lo que conduce a una sobrecarga en órganos como hígado y páncreas18. Entre un 25 y 60% de los pacientes con hemocromatosis desarrollan DM2. En modelos de hemocromatosis en ratón, se ha encontrado un aumento en el estrés oxidativo de la célula β, con una disminución en la capacidad de secretar insulina26.
En estudios epidemiológicos, se ha encontrado una relación entre niveles de ferritina corporal y el desarrollo de DM2. Así por ejemplo, en el estudio de las Enfermeras (NHS), el Odd Ratio (OR) para DM2 en los quintiles extremos de ferritina (quintil 5 vs quintil 1) fue de 2,85 (p < 0,001), resultado que permaneció significativo después de ajustar por PCR27. Forouhi et al28, reportó en su estudio de caso y control anidado niveles de ferritina más altos en los casos que en los controles (hombres: 96,6 vs 67,8 ng/ml; p < 0,001, mujeres: 45,9 vs 34,8 ng/ml; p = 0,005) y un OR de 7,4 en los que se encontraban en los cuartiles más altos de ferritina comparado con el cuartil más bajo. Aunque, después de ajustar por PCR los resultados no cambiaron, sí lo hicieron cuando se ajustó por enzimas hepáticas, lo que sugiere que el Fe podría aumentar el riesgo de DM2 a través de la alteración del funcionamiento hepático.
Cabe destacar, que los estudios mencionados fueron conducidos con población aparentemente sana y que los niveles de ferritina eran mucho más bajos que aquellos observados en sujetos con hemocromatosis, como se muestra en la Figura 2.
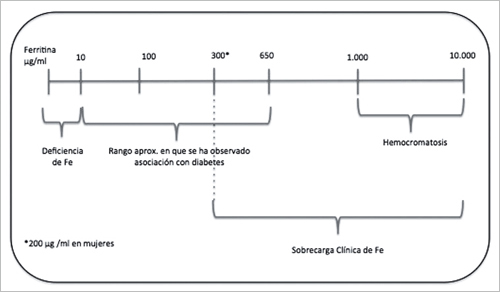
Figura 2. Rangos de ferritina asociados al riesgo de DMT2.
Potenciales mecanismos asociados al desarrollo de DM2 y hierro
Como ya se mencionó, el Fe es un potente pro-oxidante que cataliza varias reacciones que desencadenan la formación de ROS14. Es probable que el estrés oxidativo crónico catalizado por Fe en el hígado, músculo y tejido adiposo cause una respuesta inflamatoria e insulino resistencia en estos tejidos. Cabe destacar, que la inflamación puede afectar el metabolismo del Fe, es así como altos niveles de interleuquina- 6 (IL-6) e IL-1β aumentan la síntesis de ferritina y de la hormona Hepcidina, provocando redistribución del Fe corporal18.
La sobrecarga de Fe en el músculo resulta en un cambio significativo en el metabolismo de la glucosa, induciendo la disminución de su captación y oxidación, aumentando la oxidación de ácidos grasos, todo combinado con un aumento de la síntesis de glucosa en el hígado, debido a que éste órgano pierde la capacidad de extracción de insulina29. Como consecuencia, ocurre un progresivo aumento de la glucosa circulante y de la insulina circulante, aumentando el riesgo de generar glucotoxicidad.
Por otra parte, el aumento de glucosa circulante produce un cambio conformacional en la transferrina, glicosilando la proteína, lo que provoca la disminución de la afinidad de esta proteína con el Fe, permitiendo que aumente la concentración de Fe libre y por lo tanto, con una capacidad redox aumentada16.
En el tejido adiposo el exceso de Fe provoca un aumento en la lipólisis, lo que induce al incremento de ácidos grasos libres circulantes constituyéndose en un factor de riesgo cardiovascular y para el desarrollo de resistencia a la insulina a través de la lipotoxicidad, pero además el Fe provoca alteración en la captación de glucosa inducida por insulina en el adipocito, probablemente estos efectos son secundarios al estrés oxidativo provocado por el Fe, en especial la lipoperoxidación y la activación de vías como la JNK, quinasa que provoca la fosforilación en serina del receptor de insulina30.
En el páncreas, el Fe produce disfunción de la célula β a través de varios mecanismos, todos relacionados con el estrés oxidativo y con la inducción de apoptosis de la célula pancreática. En condiciones de hiperglicemia la mitocondria aumenta la generación de anión superóxido, que en presencia de Fe2+ produce el aumento de radicales hidroxilos: Si estos no son rápidamente eliminados, lo que en el páncreas es limitado debido a su disminuida capacidad antioxidante, provocan la fragmentación del DNA, la peroxidación lipídica y la activación de vías pro-apoptóticas31
La glucolipotoxicidad es la principal vía de disfunción de las células ß, el exceso de glucosa tiene distintos orígenes, en primer lugar la pérdida de respuesta a la insulina del hígado lo que provoca una continua salida de glucosa de éste órgano, por otra parte, la disminución de entrada de la glucosa al músculo y al tejido adiposo. El aumento de glucosa en el páncreas en conjunto con depósitos aumentados de Fe en éste órgano conduce en una primera etapa a la alteración de la expresión de genes como el de la insulina y si el estímulo continua en el tiempo ocurre la activación de vías apoptóticas como la vía de la JNK y AMPK que aumentan el estrés del retículo endoplasmático induciendo la apoptosis de la célula ß32.
Conclusiones
Niveles aumentados de Fe corporal medidos en forma de ferritina han mostrado en estudios epidemiológicos que constituye un factor de riesgo para el desarrollo de diabetes. El principal mecanismo que se ha propuesto, es a través del estrés oxidativo lo que conduce a un aumento de la neoglucogénesis hepática, resistencia a la insulina en el adipocito y el tejido muscular. El aumento de glucosa y de ácidos grasos circulantes induce glucolipotoxicidad en la célula ß pancreática, con la pérdida funcional en primera instancia de la secreción de insulina y finalmente con la pérdida de la célula ß a través de mecanismos de apoptosis. El hierro es un nutriente esencial para múltiples funciones orgánicas, por lo tanto, no puede ser eliminado de la dieta, sin embargo dada la evidencia existente hasta el momento del rol que tiene en el desarrollo de enfermedades crónicas como la diabetes mellitus, es necesario clarificar en primer lugar si los niveles de ferritina observados en los sujetos diabéticos son producto de una sobrecarga de Fe o más bien es el reflejo del estado pro-inflamatorio, y por lo tanto es necesario conocer como el hierro interactúa con la inflamación subyacente en las enfermedades crónicas sobretodo en obesidad dada la intima relación entre metabolismo de hierro-obesidad-inflamación.
Referencias bibliográficas
- Rajpathak SN, Crandall JP, Wylie-Rosett J, Kabat GC, Rohan TE, Hu FB. 2009. The role of iron in type 2 diabetes in humans. Bioch Bioph Acta 1790: 671-681.
- Silva M, Bonomo L, Oliveira R, de Lima W, Silva M, Pedrosa M. 2011. Effects of the interaction of diabetes and iron supplementation on hepatic and pancreatic tissues, oxidative stress markers, and liver peroxisome proliferator-activated receptor-α expression. J Clin Biochem Nutr 49: 102-108.
- McClain DA, Abraham D, Rogers J. 2006. High prevalence of abnormal glucose homeostasis secondary to decreased insulin secretion in individual with hereditary haemochromatosis. Diabetologia 49: 1661-1669.
- Ford ES, Cogswell ME. 1999. Diabetes and serum ferritin concentration among U.S.adults, Diabetes Care 22: 1978-1983.
- Ramakrishnan U, Kuklina E, Stein AD. 2002. Iron stores and
cardiovascular disease risk factors in women of reproductive age
in the United States. Am J Clin Nutr. 76: 1256-1260.
- Sheu WH, Chen YT, Lee WJ, Wang CW, Lin LY. 2003. A
relationship between serum ferritin and the insulin resistance
syndrome is present in non-diabetic women but not in non-diabetic
men. Clin Endocrinol (Oxf) 58: 380-385.
- Williams MJ, Poulton R, Williams S. 2002. Relationship of serum
ferritin with cardiovascular risk factors and inflammation in young
men and women. Atherosclerosis 165: 179-184.
- Gillum RF. 2001. Association of serum ferritin and indices of body fat distribution and obesity in Mexican American men-the third national health and nutrition examination survey. Int J Obes Relat Metab Disord 25: 639-645.
- Panel on Micronutrients food and Nutrition Board Dietary
reference intakes of vitamin A, vitamin K, arsenic, boron,
chromium, copper, iron, iodine, manganese, silicon, vanadium and
zinc. National Academy Press, Washington DC, 2001.
- Evstatiev R, Gasche C. 2011. Iron sensing and signalling. Gut doi: 10.1136/gut.2010.214312.
- Anderson GJ, Frazer DM, McLaren GD. 2009. Iron absorption and
metabolism. Curr Opin Gastroenterol 25: 129-135.
- West A, Oates P. 2008. Mechanisms of heme iron absorption:
currents questions and controversies 14: 4101-4110.
- Qiu A, Jansen M, Sakaris A. 2006. Identification of an intestinal
folate transporter and the molecular basis for hereditary folate
malabsorption. Cell 127: 917-928.
- Lipinski B. 2011. Hydroxyl radical and its scavengers in health and disease. Oxid Med Cell Longev. doi:10.1155/2011/809696.
- Lee DH, Liu DY, Jacobs Jr DR, Shin HR, Song K, Lee IK, Kim B, Hider RC. 2006. Common presence of non-transferrin-bound iron among patients with type 2 diabetes, Diabetes Care 29:1090-1095.
- Fernández-Real JM, Moreno JM, López-Bermejo A, Chico B, Vendrell J, Ricart W. 2007. Circulating soluble transferrin receptor according to glucose tolerance status and insulin sensitivity. Diabetes. Care 30: 604-608.
- Jehn M, Clark J, Guallar E. 2004. Serum ferritin and risk of metabolic síndrome in US adults. Diabetes Care 27: 2422-2428.
- Ganz T, Nemeth E. 2011. Hepcidin and disorders of iron metabolism. Annu Rev Med 62: 347-360.
- Pastor N, Weinstein H, Jamison E, Brenowitz M. 2000. A detailed interpretation of OH radical footprints in a TBP-DNA complex reveals the role of dynamics in the mechanism of sequencespecific binding. J Mol Biol 34: 55-68.
- Poulsen HE, Specht E, Broedbaek K, Henriksen T, Ellervik C, Mandrup-Poulsen T, et al. 2012. RNA modifications by oxidation: A novel disease mechanism? Free Radic Biol Med 15: 1353-1361.
- Cooksey RC, Jouihan HA, Ajioka RS, Hazel MW, Jones DL, Kushner JP, et al. 2004. Oxidative stress, β-cell apoptosis, and decreased insulin secretory capacity in mousemodels of hemochromatosis. Endocrinology 145: 5305-5312
- Tuomainen TP, Nyyssönen K, Salonen R, Tervahauta A, Korpela H, Lakka T, et al. 1997. Body iron stores are associated with serum insulin and blood glucose concentrations. Population study in 1.013 eastern Finnish men. Diabetes Care 20: 426-428.
- Fumeron F, Pean F, Driss F, Balkau B, Tichet J, Marre M, et al. 2006. Ferritin and transferrin are both predictive of the onset of hyperglycemia in men and women over 3 years: the data from an epidemiological study on the Insulin Resistance Syndrome (DESIR) study, Diabetes Care 29: 2090-2094.
- Gillum RF. 2001. Association of serum ferritin and indices of body fat distribution and obesity in Mexican American men-the third national health and nutrition examination survey, Int J Obes Relat Metab Disord 25: 639-645.
- Fernández-Real JM, Vendrell J, Baiget M, Gimferrer E, Ricart W. 1999. C282Y and H63D mutations of the hemochromatosis candidate gene in type 2 diabetes, Diabetes Care 22: 525-526.
- Huang J, Jones D, Luo B, Sanderson M, Soto J, Abel ED, et al. 2011. Iron overload and diabetes risk: A shift from glucose to fatty acid oxidation and increased hepatic glucose production in a mouse model of hereditary hemochromatosis. Diabetes 60: 80-88.
- Jiang R, Manson JE, Meigs JB, Ma J, Rifai N, Hu FB. 2004. Body iron stores in relation to risk of type 2 diabetes in apparently healthy women, JAMA 291: 711-717.
- Forouhi NG, Harding AH, Allison M, Sandhu MS, Welch A, Luben R, et al. 2007. Elevated serum ferritin levels predict new onset type 2 diabetes: results from the EPIC-Norfolk prospective study. Diabetologia 50: 949-956.
- Ferrannini E. 2000. Insulin resistance, iron, and the liver. Lancet 355: 2181-2182.
- Green A, Basile R, Rumberger JM. 2006. Transferrin an iron induce insulin resistance of glucose transport in adipocytes. 55: 1042-1045.
- Ma ZA, Zhao Z, Turk J. 2012. Mitochondrial dysfunction and β-cell failure in type 2 diabetes mellitus. Exp Diab Res 2012 doi:10.1155/2012/703538.
- Chen J, Saxena G, Mungrue IN. 2008. Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes 57: 938-944.