Hiperaldosteronismo primario
Carlos Fardella B., Lorena Mosso G. y Cristian Carvajal M.a
Primary aldosteronism
Departamento de Endocrinología, Facultad de Medicina, Pontificia Universidad Católica de Chile. Santiago de Chile.
aBioquímico
Trabajo financiado por proyectos Fondecyt 1011035 y 1070876
Correspondencia a: Dr. Carlos Fardella B. Departamento de Endocrinología, Facultad de Medicina, Pontificia Universidad Católica de Chile. Lira 85, 5o piso. Santiago, Chile. Fono: 354-3095. Fax: 638-5675. E mail: cfardella@med.puc.cl
Recibido: 17 Marzo de 2009
Aceptado: 05 Junio de 2009
Primary aldosteronism (PA) is a known cause of hypertension. In the kidney, aldosterone promotes sodium and water reabsorption, increasing the intravascular volume and blood pressure (BP). In the cardiovascular system, aldosterone modifies endotelial and smooth muscle cell response, increasing cardiovascular risk in a blood pressureindependent way. Recently, a high prevalence of PA (near to 10%) in hypertensive population, has been detected measuring plasma aldosterone/renin activity ratio (ARR) as screening test. This ratio increases along with the severity of the hypertensive disease. The diagnostic work up of PA should confirm the autonomy of aldosterone secretion from the renin-angiotensin system and should differentiate the clinical subtypes of the disease. These are idiopathic aldosteronism (IA) and aldosterone-producing adenoma (APA). Other causes are familial hyperaldosteronism (FH) type I (glucocorticoid-remediable aldosteronism), FH-II (non glucocorticoid-remediable aldosteronism), primary adrenal hyperplasia and adrenal carcinoma. This article reviews the prevalence, diagnosis and treatment of PA and also the clinical, biochemical and genetic characteristics of its different subtypes.
Key words: Aldosterone; Hyperaldosteronism; Hypertension.
Reproducido con autorización de la Revista Médica de Chile (Rev. Méd. Chile 2008; 136: 905-914).
La hipertensión arterial (HTA) es una de las patologías más frecuentes en la población general y se estima que afecta aproximadamente a 26% de la población mundial1. En Chile, de acuerdo a la última Encuesta Nacional de Salud, la prevalencia actual de HTA es 33% (ENS 2006, Ministerio de Salud, Chile). Debido a los cambios en los perfiles epidemiológicos actuales, la prevalencia mundial de HTA podría aumentar a 30% en 2025 (1,5 billones de personas)1. En los pacientes hipertensos existe una mayor morbimortalidad por accidentes cerebrovasculares, síndromes coronarios agudos e insuficiencia cardíaca. El hiperaldosteronismo primario (HAP) es una de las causas conocidas de hipertensión arterial. En estos casos, la HTA es secundaria a una producción excesiva y autónoma de aldosterona, que a nivel renal induce un aumento en la reabsorción de sal y agua, lo que se traduce en un aumento del volumen intravascular y, secundariamente, en elevación de la presión arterial2. Tradicionalmente la prevalencia del HAP ha sido estimada en menos de 1% de los hipertensos cuando la hipokalemia es usada como test de screening3-5. Sin embargo, estudios recientes han demostrado que el HAP puede ser mucho más prevalente cuando se miden aldosterona plasmática (AP), la actividad de renina plasmática (ARP) y la relación AP/ARP en el screening de esta enfermedad. En el presente milenio, múltiples estudios han usado la determinación de la relación AP/ARP como screening de HAP y han usado el test de supresión con fludrocortisona (TSF) o sobrecarga salina (TSS) para cofirmar el diagnóstico6-18. Los resultados de estos estudios demuestran que la prevalencia de HAP alcanza cifras cercanas a 5%-20% de la población de hipertensos19. Estas cifras son más altas cuando se consideran hipertensos más severos (estados 2 y 3 del JNC-VI), donde las cifras pueden elevarse hasta 15% y en pacientes refractarios a terapia antihipertensiva donde la prevalencia puede llegar hasta 20% de la población estudiada15-19. Además, estos estudios han demostrado que la minoría (30%) de los casos presentan hipokalemia, por lo que se ha acuñado el término de HAP normokalémico para identificar esta entidad. Estas evidencias han llevado a plantear que el HAP sería una condición patológica continua en la cual sólo una minoría de los sujetos afectados presentaría el cuadro clínico clásico con hipokalemia20,21.
La importancia de diagnosticar el HAP ha tomado relevancia en los últimos años no sólo por su alta prevalencia sino también por los efectos deletéreos en varios órganos, como ocurre en el corazón y vasos sanguíneos vía receptores no epiteliales e independientes de los cambios en la presión arterial. Este efecto deletéreo en el sistema cardiovascular incluye fibrosis miocárdica, reducción de la fibrinolisis y disfunción endotelial. El estudio RALES (Randomized Aldactone Evaluation Study) y el EPHESUS (Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study) demostraron que la adición de un antagonista de aldosterona a la terapia óptima, reducía la mortalidad en 30% en pacientes con falla cardiaca tipo IV y reducía la mortalidad en 15% en pacientes con disfunción ventricular izquierda después de un infarto agudo al miocardio (IAM), respectivamente22,23. Por su parte, los pacientes con HAP tienen mayor riesgo de eventos cardiovasculares que los esperados para sus niveles de presión arterial, mayor hipertrofia ventricular izquierda, accidente vascular encefálico (AVE) e IAM que los pacientes hipertensos esenciales24. Recientemente, la aldosterona ha sido involucrada también como factor de riesgo independiente de la presión arterial para el síndrome metabólico25. De particular interés son los datos que apuntan a que inhibidores de la aldosterona pueden retardar la aparición de diabetes en hipertensos26. Estos datos apuntan a un nuevo rol de la aldosterona frente al metabolismo de los hidratos de carbono. El rol de la aldosterona en la progresión de la enfermedad cardiovascular y renal se está clarificando recientemente, es así que altos niveles de aldosterona combinados con una dieta rica en sal inducen una respuesta vascular inflamatoria independiente de la elevación de la presión arterial. Esta respuesta se caracteriza por infiltración perivascular de leucocitos, remodelación vascular con necrosis fibrinoide de la media y secundariamente una isquemia y alteración necrótica en el tejido comprometido. En pacientes con HAP se han observado hallazgos similares, en particular una patología vascular necrotizante con una marcada infiltración leucocitaria inflamatoria perivascular. Estos efectos no epiteliales de la aldosterona, producida en forma sistémica o localmente, se reducen por administración de bloqueadores específicos de aldosterona, los cuales disminuyen los marcadores séricos del recambio de colágeno, y secundariamente la morbimortalidad. La aldosterona puede aumentar la presión sanguínea, aumentando la resistencia vascular, actuando sobre el endotelio vascular y las células del músculo liso. En células de la vasculatura, la aldosterona induce una variedad de efectos rápidos que contribuyen a modificar la resistencia vascular, como la elevación de la concentración de calcio intracelular, los niveles de inositol trifosfato (IP3) y cAMP. En el riñón, la aldosterona induce también un aumento en la resistencia vascular y en la presión capilar glomerular (arteriolas aferentes y eferentes) a través de la activación de la fosfolipasa C con una activación subsecuente de los canales de calcio dependientes de voltaje. Estas acciones vasoconstrictoras en la microcirculación glomerular juegan un importante rol en la fisiopatología y progresión de la enfermedad renal2,27,28. En relación a la etiología del HAP, los subtipos más prevalentes son la hiperplasia adrenocortical bilateral o hiperaldosteronismo idiopático y el adenoma productor de aldosterona. Otras causas son el hiperaldosteronismo familiar tipo I (HF-I) (aldosteronismo remediable por glucocorticoides) y el HFII (aldosteronismo no remediable por glucocorticoides), la hiperplasia adrenal primaria y el carcinoma suprarrenal19.
Regulación de la secreción de aldosterona
La biosíntesis de aldosterona está controlada por el citocromo P450c11AS (aldosterona sintetasa), el cual convierte en pasos sucesivos la 11-deoxicorticosterona en aldosterona. Este citocromo es codificado por el gen CYP11B2 el cual es regulado por angiotensina II y potasio vía proteína kinasa C. Esta enzima es diferente del citocromo P450c11ß (11ß-hidroxilasa), codificado por el gen CYP11B1 el cual es expresado en la zona fasciculata y es el encargado de convertir el 11-deoxicortisol a cortisol (Figura 1). Los genes CYP11B1 y CYP11B2 presentan una alta homología, con 90% de identidad en las secuencias intrónicas y 95% en las secuencias exónicas. Además, ambos están localizados en el brazo largo del cromosoma 829.
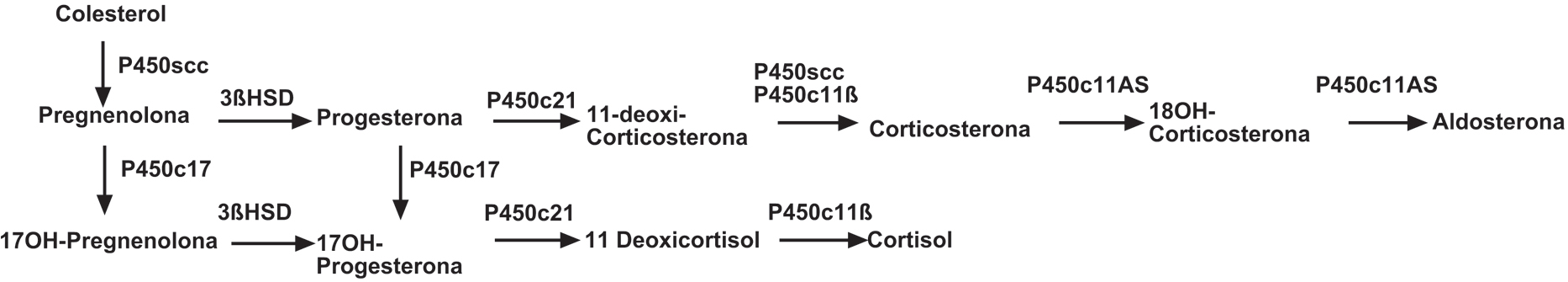
Figura 1. Vías de síntesis de esteroides humanos suprarrenales, aldosterona y cortisol. La síntesis de aldosterona, es llevada a cabo en la zona glomerulosa de la corteza suprarrenal y su etapa limitante es la reacción catalizada por la enzima aldosterona sintetasa (P450c11AS).
En el HAP, la producción de aldosterona es autónoma y no es controlada por angiotensina II, induciendo una supresión del eje renina-angiotensina-aldosterona (RAA)30-32. Esta autonomía es definida por la imposibilidad de frenar la producción de aldosterona con maniobras que normalmente suprimen su producción. La respuesta a estímulos que normalmente activan (postura erguida) el sistema RAA es variable. En adenomas productores de aldosterona (APA) generalmente no hay respuesta, pero en el hiperaldosteronismo idiopático (HAI) la producción de aldosterona usualmente aumenta30-33.
En pacientes con hiperaldosteronismo familiar tipo I (HF-I) la producción de aldosterona está bajo control de la hormona adrenocorticotrofina (ACTH), por lo cual los pacientes afectados pueden ser tratados con glucocorticoides. El HF-I es causado por una recombinación desigual entre los genes que codifican para la 11ß-hidroxilasa (CYP11B1) y la aldosterona sintetasa (CYP11B2), resultando en un gen quimérico el cual tiene actividad aldosterona sintasa pero es regulado por ACTH33,34. Este gen quimérico contiene en su porción amino terminal 3’ los elementos que determinan la respuesta a ACTH fusionado a las secuencias codificadoras del gen CYP11B2. Este gen es expresado en la zona fasciculata y determina la sobreproducción de aldosterona y de los esteroides adrenales 18-hidroxicortisol y 18-oxocortisol, los cuales se encuentran bajo control de ACTH y por tanto son supresibles con glucocorticoides. En pacientes con APA la secreción de aldosterona es dependiente al menos en parte de ACTH. Este hecho podría ser explicado por un aumento en la expresión de receptores para ACTH encontrado en algunos pacientes con adenoma. Por el contrario, los pacientes con HAI presentan una muy escasa o nula dependencia al estímulo de ACTH.
El fenotipo del hiperaldosteronismo familiar tipo II (HF-II) es clínicamente indistinguible de HAP esporádico, respecto a la edad de diagnóstico, sexo, frecuencia de hipokalemia, AP, ARP35. El HF-II puede presentarse como una hiperplasia adrenocortical bilateral o como un adenoma. El modo de herencia del HF-II se mantiene especulativo, pero la transmisión vertical sugiere una herencia dominante autosómica. Varios genes candidatos han sido estudiados, por ejemplo el gen del receptor de angiotensina II tipo 1 y el CYP11B2 “aldosterona sintetasa”; sin embargo, ambos genes han sido excluidos por análisis de linkage. Recientemente ha concluido una amplia búsqueda genómica, la cual determinó que existe un locus en el cromosoma 7 (cr 7p22) asociado a HF-II. Los genes candidatos localizados en esta región corresponden a genes involucrados en tumorogénesis o que tienen un posible rol en la regulación del sistema RAA; éstos serían: GPR30, un receptor acoplado a proteína G; PMS2; PRKAR1B, una proteína reguladora de la proteína kinasa dependiente de cAMP; y el gen centaurinaalfa 12. PRKAR1B es el candidato favorito, pero a la fecha no existe evidencia significativa que apoye esta hipótesis36. La determinación del gen causante del HF-II puede estar implicado en el HAP esporádico y la hipertensión, asimismo ayudará a conocer mejor la regulación de la fisiopatología de la hipertensión arterial.
Diagnóstico de hiperaldosteronismo primario
El diagnóstico de hiperaldosteronismo primario está enfocado a confirmar la autonomía de la secreción de aldosterona del eje renina angiotensina. Un esquema utilizado para realizar el diagnóstico de hiperaldosteronismo primario se muestra en la Figura 2.
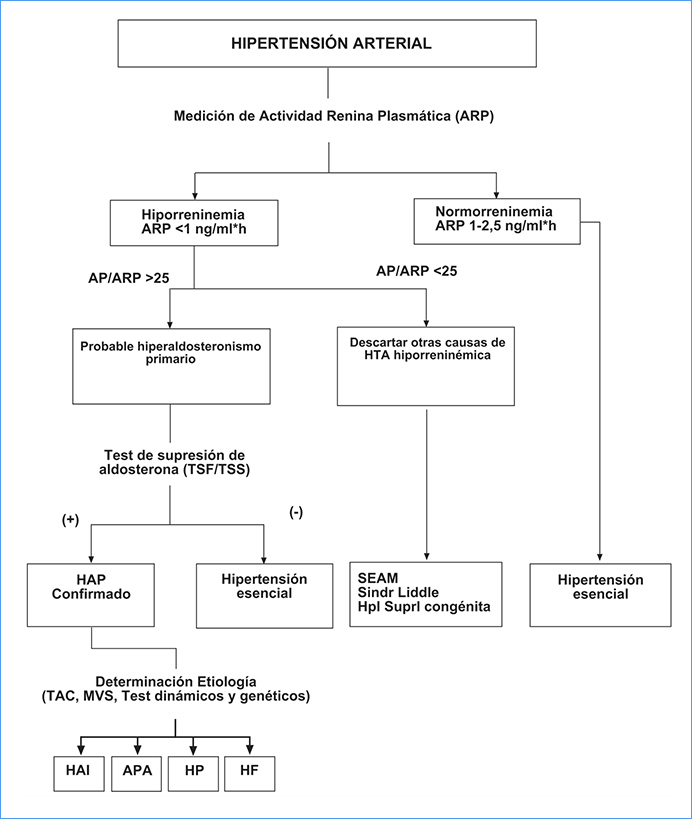 Figura 2. Diagrama de flujo diagnóstico para el hiperaldosteronismo primario y diagnóstico diferencial (HAI: HA idiopático; APA: adenoma productor de aldosterona; HP: hiperplasia primaria; HF: HA familiar; SEAM: síndrome de exceso aparente de mineralocorticoides; TAC: tomografía axial computada; MVS: muestreo de venas suprarrenales).
Figura 2. Diagrama de flujo diagnóstico para el hiperaldosteronismo primario y diagnóstico diferencial (HAI: HA idiopático; APA: adenoma productor de aldosterona; HP: hiperplasia primaria; HF: HA familiar; SEAM: síndrome de exceso aparente de mineralocorticoides; TAC: tomografía axial computada; MVS: muestreo de venas suprarrenales).1. Screening del hiperaldosteronismo primario
a) Potasio. La hipokalemia ha sido considerada como el elemento clásico para el diagnóstico de HAP. Sin embargo, estudios recientes han demostrado que niveles disminuidos de potasio plasmático se presentan sólo en 20% de los pacientes afectados por un HAP7-9. El potasio plasmático puede ser influenciado por la severidad y duración del hiperaldosteronismo, la ingesta de sodio y la sensibilidad de los túmulos renales a la aldosterona. De esta forma, el HAP normokalémico constituye la forma más común de presentación de la enfermedad, y la variante hipokalémica probablemente la forma más severa de la enfermedad.
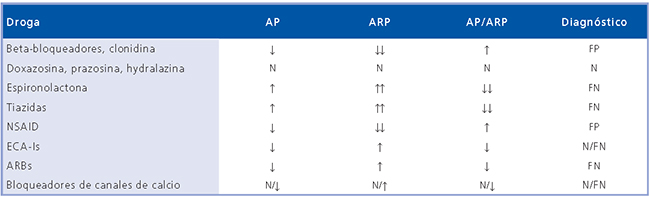
b) Determinación de la relación AP/ARP. La determinación aislada de aldosterona plasmática (AP) o actividad de renina plasmática (ARP) no es suficiente para el diagnóstico de HAP, en ambos casos hay condiciones que pueden inducir a errores en la determinación. Este margen de error puede ser disminuido si se calcula la relación AP/ARP37. Así, en el diagnóstico de HAP la pista más importante es demostrar un valor suprimido de ARP y un valor elevado de la relación AP/ARP. Recientemente nuestro grupo validó en normotensos un valor normal para la relación AP/ARP < 24, motivo por el cual consideramos elevada toda relación igual o superior a 257,38. Además establecimos que una relación AP/ARP elevada podía dar una falsa sospecha de HAP cuando valores normales de AP eran amplificados varias veces por valores muy bajos de ARP. Por esta razón, propusimos que el límite inferior de ARP no debía ser menor de 0,3 ng/mL*h cuando se realiza el cálculo. En pacientes con una relación de AP/ ARP mayor que 50, y niveles elevados de AP el diagnóstico de HAP es casi siempre confirmado, independiente del límite inferior de ARP considerado. Una ventaja de la determinación de la relación AP/ARP es que se ve menos afectada por la administración de drogas y por la posición de los sujetos, permitiendo su uso bajo condiciones menos restrictivas37,39. A pesar de esto, debe tenerse presente que drogas como los diuréticos y los bloqueadores del receptor de angiotensina pueden dar falsos negativos y el propanolol falsos positivos (Tabla 1). Sin embargo, si en un paciente que está tomando diuréticos se detecta una elevación de la relación AP/ARP, este hecho es fuertemente sugerente de un HAP. En nuestra experiencia, las muestras de AP y ARP pueden ser tomadas entre las 8 y las 10 AM después de que el paciente ha permanecido sentado por 15 min, y no necesariamente después de estar acostado por 2 h como indicaba el protocolo original40.
Tabla 1. Efecto de drogas antihipertensivas en los niveles plasmáticos de aldosterona y renina
2. Test confirmatorios de hiperaldosteronismo primario
a) Test de supresión con fludrocortisona (TSF). La confirmación del diagnóstico de HAP se basa en demostrar la autonomía de la producción de aldosterona a través de un test de supresión. En el test de fludrocortisona los niveles de AP son medidos en la condición basal y después de 4 días de administrar acetato de fludrocortisona (0,4 mg/día) bajo una dieta suplementada con sodio, 110 mmol/día32,41. Las muestras de sangre se toman al quinto día a las 08:00 AM. El test de fludrocortisona es considerado positivo cuando la AP mantiene valores sobre 5 ng/dl42,43.
b) Test de sobrecarga salina (TSS). Una alternativa al test de fludrocortisona, es el test de sobrecarga o infusión salina. Consiste en la administración de una solución salina isotónica de 500 ml/h durante dos a cuatro horas42,44. La persistencia de niveles de AP sobre 5 ng/dl confirma el diagnóstico de HAP. Los autores recomiendan que ambos test sean cuidadosamente monitorizados en ancianos y evitados en pacientes con hipertensión severa, insuficiencia cardíaca severa, accidente vascular o infarto del miocardio.
Clasificación de los subtipos de hiperaldosteronismo primario
Una vez que el diagnóstico de HAP ha sido confirmado, es necesario determinar la etiología del HAP, lo cual es importante para tomar decisiones terapéuticas.
1. Test postural y test de infusión de angiotensina II. En pacientes de HAI la aldosterona generalmente aumenta con los cambios de postura, porque el sistema reninaangiotensina no está totalmente suprimido. Por el contrario, en pacientes con APA los niveles de aldosterona generalmente disminuyen en paralelo con la secreción circadiana de cortisol. La exactitud de estos tests es aumentada con la medicion simultanea de los niveles plasmaticos de cortisol. El valor predictivo del test postural en distinguir entre HAI y APA es cercano a 90%. Sin embargo, existen reportes de que pacientes con APA pueden responder al test postural y a la infusion de angiotensina 242,45 y que pacientes con hiperplasia adrenal primaria pueden no presentar respuesta al test postural43,45. Mas aun, pacientes con HF-I muestran una disminucion en la aldosterona similar a la que presentan pacientes con APA.
2. Test de supresion con dexametasona. Este test ha sido usado tradicionalmente en el diagnostico de HF-I dado que la produccion de aldosterona esta bajo control de ACTH46. En este test se realizan mediciones basales de aldosterona y cortisol, y despues de 2 a 4 dias de ingerir dexametasona (2,0 mg/dia). El test se considera positivo cuando los niveles de aldosterona se mantienen sobre los 4 ng/dl47,48. Para asegurar la confiabilidad del test es necesario confirmar la supresion del cortisol plasmatico a menos de 2,5 μg/dl. Sin embargo, comunicaciones recientes han demostrado que este test tiene una alta frecuencia de falsos positivos7,48 cuando es comparado con el test genetico usado en el diagnostico de pacientes con HF-I49.
3. Determinacion de 18 hidroxicortisol y 18 oxocortisol. Niveles elevados de 18-hidroxicortisol y 18-oxocortisol se encuentran en pacientes con HF-I, APA e HP, pero no en pacientes con HAI50. En pacientes con HF-I la elevacion puede ser mas de 10 veces el valor normal, en cambio en APA y HP la elevacion es discreta.
4. Test genetico para HF-I. En la actualidad, es posible detectar la presencia del gen quimerico CYP11B1/ CYP11B2, el cual es una herramienta diagnostica para HF-I34,51,53. La deteccion de este gen fue realizada inicialmente utilizando la tecnica de Southern blotting, pero en el ultimo tiempo ha sido realizada con exito usando la tecnica de longextension PCR, tambien llamado XL-PCR52,53.
Procedimientos de localizacion
Los procedimientos de localizacion deben ser llevados a cabo solo despues que el diagnostico de HAP ha sido establecido.
1. Tomografia axial computada (TAC). La TAC es un procedimiento muy util para detectar la mayoria de los adenomas, aun cuando estos sean menores de 5 mm. En pacientes con HAI las glandulas suprarrenales aparecen crecidas en forma bilateral o pueden tambien aparecer de tamano normal. Aunque no hay una medicion exacta del tamano de la glandula suprarrenal, la TAC es considerada anormal cuando cualquier area de ella es mayor de 10 mm54. En cambio, los adenomas aparecen como una masa unilateral de baja densidad, generalmente menores de 2 cm de diametro54. La deteccion de un nodulo mayor de 6 cm podria hacer sospechar la presencia de un carcinoma suprarrenal55. La mas clara desventaja de la TAC es cuando la bioquimica de HAP no es claramente definida, y la presencia de un incidentaloma podria ser erroneamente confundida con un adenoma. Mas aun, una hiperplasia micromacro nodular con un nodulo dominante podria llevar a un falso diagnostico de adenoma56. La experiencia con resonancia nuclear magnetica (RNM) no parece ofrecer ventajas sobre la TAC.
2. Muestreo de venas suprarrenales (MVS). Es considerado el metodo mas confiable para probar lateralizacion como ocurre en los casos de adenoma o hiperplasia (HP)57. El procedimiento se realiza por via femoral y se cateterizan ambas venas suprarrenales y la vena cava inferior. Se considera que existe lateralizacion cuando la relacion aldosterona/ cortisol en un lado es al menos 2 veces mayor que la determinada en la vena cava, a diferencia del lado contralateral donde la relacion es semejante en ambos sitios. Este metodo requiere considerable experiencia del radiologo y tiene riesgo de hemorragia suprarrenal.
Terapia del hiperaldosteronismo primario
El objetivo del tratamiento es prevenir la morbimortalidad asociada con la hipertensión, las alteraciones hidroelectrolíticas y el daño cardiovascular. La aproximación terapéutica del HAP depende del subtipo etiológico. Así, la cirugía es el tratamiento de elección en pacientes con APA y HP58. En la actualidad la cirugía laparoscópica es de elección dado que presenta menos complicaciones y los períodos de hospitalización y recuperación son más cortos. La corrección quirúrgica del HAP generalmente normaliza o disminuye significativamente las cifras de presión arterial59. La persistencia de la hipertensión puede estar relacionada con la severidad o cronicidad de la enfermedad hipertensiva o por la coexistencia de hipertensión esencial. El tratamiento medicamentoso es la terapia de elección para pacientes afectados por HAI. La espironolactona, un antagonista de la aldosterona a nivel de su receptor, ha sido la droga tradicionalmente usada. Las dosis varían entre 25- 100 mg/día, con lo cual se alcanza un efectivo control de la presión arterial y de la hipokalemia en la mayoría de los casos58. Sin embargo, su uso produce efectos adversos como ginecomastia, disfunción eréctil, disminución de la libido, síntomas gastrointestinales e irregularidades menstruales. La eplerenone es un nuevo antagonista selectivo del receptor de mineralocorticoides, recientemente aprobado para el tratamiento de la hipertensión arterial. La ventaja con respecto a la espironolactona es que la eplerenona no presenta los efectos adversos descritos para la espironolactona y, por ende, aparece como una droga de elección para el manejo de estos pacientes. Otras alternativas de tratamiento son el amiloride y el triamterene, drogas que impiden la acción de la aldosterona al inducir un bloqueo del canal de sodio a nivel renal y con ello impiden la retención de sodio y la pérdida de potasio60. El nifedipino, un bloqueador de los canales de sodio, también ha demostrado ser efectivo en el control de la presión arterial, pero los resultados a largo plazo han sido poco alentadores. En los pacientes con un HF-I el tratamiento de elección es la dexametasona a bajas dosis entre 0,125-0,5 mg/día61. Sin embargo, también pueden responder a prednisona o hidrocortisona. En niños es recomendable ajustar la dosis por superficie corporal, para evitar una sobre dosificación.
Conclusiones
El hiperaldosteronismo constituye la forma más prevalente de hipertensión arterial secundaria. Además el hiperaldosteronismo ha cobrado relevancia al demostrarse que la aldosterona per se puede ejercer un efecto deletéreo directo en varios órganos, independiente del aumento de la presión arterial. El screening debería realizarse en hipertensos moderados, severos o resistentes a terapia usando la relación aldosterona/renina y no la medición del potasio plasmático. La sospecha de hiperaldosteronismo debe certificarse usando tests confirmatorios (fludrocortisona o sobrecarga salina). Una vez confirmada su existencia deben usarse procedimientos que permitan su clasificación para orientar la terapia (médica o quirúrgica). La detección de formas hereditarias de la enfermedad abre un campo de investigación que no sólo permitirá entender mejor la fisiopatología de la enfermedad sino que también permitirá a futuro el consejo genético en familias afectadas.
Referencias
- KEARNEY PM, WHELTON M, REYNOLDS K, MUNTNER P, WHELTON PK, HE J. Global burden of hypertension: analysis of worldwide data. Lancet 2005; 365: 217-23.
- JACKSON RV, LAFFERTY A, TORPY DJ, STRATAKIS C. New genetic insights in familial hyperaldosteronism. Ann N Y Acad Sci 2002; 970: 77-88.
- CONN JW, COHEN EL, ROVNER DR, NESBIT RM. Normokalemic Primary Aldosteronism. A Detectable Cause of Curable “Essential” Hypertension. JAMA 1965; 193: 200-6.
- TUCKER RM, LABARTHE DR. Frequency of surgical treatment for hypertension in adults at the Mayo Clinic from 1973 through 1975. Mayo Clin Proc 1977; 52: 549-5.
- BLUMENFELD JD, SEALEY JE, SCHLUSSEL Y, VAUGHAN ED JR, SOS TA, ATLAS SA ET AL. Diagnosis and treatment of primary hyperaldosteronism. Ann Intern Med 1994; 121: 877-85.
- MULATERO P, STOWASSER M, LOH KC, FARDELLA CE, GORDON RD, MOSSO L ET AL. Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. J Clin Endocrinol Metab 2004; 89: 1045-50.
- FARDELLA CE, MOSSO L, GÓMEZ-SÁNCHEZ C, CORTÉS P, SOTO J, GÓMEZ L ET AL. Primary hyperaldosteronism in essential hypertensives: prevalence, biochemical profile, and molecular biology. J Clin Endocrinol Metab 2000; 85: 1863-7.
- LIM PO, DOW E, BRENNAN G, JUNG RT, MACDONALD TM. High prevalence of primary aldosteronism in the Tayside hypertension clinic population. J Hum Hypertens 2000; 14: 311-5.
- LOH KC, KOAY ES, KHAW MC, EMMANUEL SC, YOUNG WF JR. Prevalence of primary aldosteronism among Asian hypertensive patients in Singapore. J Clin Endocrinol Metab 2000; 85: 2854-9.
- NISHIKAWA T, OMURA M. Clinical characteristics of primary aldosteronism: its prevalence and comparative studies on various causes of primary aldosteronism in Yokohama Rosai Hospital. Biomed Pharmacother 2000; 54 Suppl 1: 83s-85s.
- RAYNER BL, OPIE LH, DAVIDSON JS. The aldosterone/renin ratio as a screening test for primary aldosteronism. S Afr Med J 2000; 90: 394-400.
- GALLAY BJ, AHMAD S, XU L, TOIVOLA B, DAVIDSON RC. Screening for primary aldosteronism without discontinuing hypertensive medications: plasma aldosterone-renin ratio. Am J Kidney Dis 2001; 37: 699-705.
- SCHWARTZ GL, CHAPMAN AB, BOERWINKLE E, KISABETH RM, TURNER ST. Screening for primary aldosteronism: implications of an increased plasma aldosterone/ renin ratio. Clin Chem 2002; 48: 1919-23.
- CALHOUN DA, NISHIZAKA MK, ZAMAN MA, THAKKAR RB, WEISSMANN P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 2002; 40: 892-6.
- MOSSO L, CARVAJAL C, GONZÁLEZ A, BARRAZA A, AVILA F, MONTERO J ET AL. Primary aldosteronism and hypertensive disease. Hypertension 2003; 42: 161-5.
- ROSSI E, REGOLISTI G, NEGRO A, SANI C, DAVOLI S, PERAZZOLI F. High prevalence of primary aldosteronism using postcaptopril plasma aldosterone to renin ratio as a screening test among Italian hypertensives. Am J Hypertens 2002; 15: 896-902.
- STOWASSER M, GORDON RD, GUNASEKERA TG, COWLEY DC, WARD G, ARCHIBALD C ET AL. High rate of detection of primary aldosteronism, including surgically treatable forms, after ‘non-selective’ screening of hypertensive patients. J Hypertens 2003; 21: 2149-57.
- OMURA M, SAITO J, YAMAGUCHI K, KAKUTA Y, NISHIKAWA T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res 2004; 27: 193- 202.
- FARDELLA C, CARVAJAL C, MOSSO L. Primary Hyperaldosteronism in the Hypertensive Disease. Curr Hypertens Reviews 2006; 2: 36-40.
- KAPLAN NM. Cautions over the current epidemic of primary aldosteronism. Lancet 2001; 357: 953-4.
- PITT B, ZANNAD F, REMME WJ, CODY R, CASTAIGNE A, PÉREZ A ET AL. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341: 709-17.
- PITT B, REMME W, ZANNAD F, NEATON J, MARTÍNEZ F, RONIKER B ET AL. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348: 1309-21.
- MILLIEZ P, GIRERD X, PLOUIN PF, BLACHER J, SAFAR ME, MOURAD JJ. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol 2005; 45: 1243-8.
- FALLO F, VEGLIO F, BERTELLO C, SONINO N, DELLA MEA P, ERMANI M ET AL. Prevalence and characteristics of the metabolic syndrome in primary aldosteronism. J Clin Endocrinol Metab 2006; 91: 454-9.
- COOPER ME. The role of the renin-angiotensinaldosterone system in diabetes and its vascular complications. Am J Hypertens 2004; 17: 16S-20S; quiz A2-4.
- ARIMA S, KOHAGURA K, XU HL, SUGAWARA A, ABE T, SATOH F ET AL. Nongenomic vascular action of aldosterone in the glomerular microcirculation. J Am Soc Nephrol 2003; 14: 2255-63.
- ARIMA S, KOHAGURA K, XU HL, SUGAWARA A, URUNO A, SATOH F ET AL. Endothelium-derived nitric oxide modulates vascular action of aldosterone in renal arteriole. Hypertension 2004; 43: 352-7.
- ROCHA R, FUNDER JW. The pathophysiology of aldosterone in the cardiovascular system. Ann N Y Acad Sci 2002; 970: 89-100.
- MORNET E, DUPONT J, VITEK A, WHITE PC. Characterization of two genes encoding human steroid 11 betahydroxylase (P-450(11) beta). J Biol Chem 1989; 264: 20961-7.
- GANGULY A, MELADA GA, LUETSCHER JA, DOWDY AJ. Control of plasma aldosterone in primary aldosteronism: distinction between adenoma and hyperplasia. J Clin Endocrinol Metab 1973; 37: 765-75.
- VALLOTTON MB. Primary aldosteronism. Part II. Differential diagnosis of primary hyperaldosteronism and pseudoaldosteronism. Clin Endocrinol (Oxf) 1996; 45: 53-60.
- VALLOTTON MB. Primary aldosteronism. Part I. Diagnosis of primary hyperaldosteronism. Clin Endocrinol (Oxf) 1996; 45: 47-52.
- GORDON RD. Primary aldosteronism. J Endocrinol Invest 1995; 18: 495-511.
- PASCOE L, CURNOW KM, SLUTSKER L, CONNELL JM, SPEISER PW, NEW MI ET AL. Glucocorticoid-suppressible hyperaldosteronism results from hybrid genes created by unequal crossovers between CYP11B1 and CYP11B2. Proc Natl Acad Sci USA 1992; 89: 8327-31.
- TORPY DJ, GORDON RD, LIN JP, HUGGARD PR, TAYMANS SE, STOWASSER M ET AL. Familial hyperaldosteronism type II: description of a large kindred and exclusion of the aldosterone synthase (CYP11B2) gene. J Clin Endocrinol Metab 1998; 83: 3214-8.
- ELPHINSTONE MS, GORDON RD, SO A, JESKE YW, STRATAKIS CA, STOWASSER M. Genomic structure of the human gene for protein kinase A regulatory subunit R1-beta (PRKAR1B) on 7p22: no evidence for mutations in familial hyperaldosteronism type II in a large affected kindred. Clin Endocrinol (Oxf) 2004; 61: 716-23.
- CORTÉS P, FARDELLA C, OESTREICHER E, GAC H, MOSSO L, SOTO J ET AL. [Excess of mineralocorticoids in essential hypertension: clinical-diagnostic approach]. Rev Méd Chile 2000; 128: 955-61.
- MCKENNA TJ, SEQUEIRA SJ, HEFFERNAN A, CHAMBERS J, CUNNINGHAM S. Diagnosis under random conditions of all disorders of the renin-angiotensin-aldosterone axis, including primary hyperaldosteronism. J Clin Endocrinol Metab 1991; 73: 952-7.
- MONTERO J, SOTO J, FARDELLA C, FORADORI A, VALDÉS G. [Measurement of low levels of plasma renin activity. A methodological improvement]. Rev Méd Chile 1998; 126: 151-4.
- STREETEN DH, TOMYCZ N, ANDERSON GH. Reliability of screening methods for the diagnosis of primary aldosteronism. Am J Med 1979; 67: 403-13.
- GILL J. Hyperaldosteronism. In: Becker K, ed. Principles and practice of endocrinology and metabolism. 2nd ed. Philadelphia: JB Lippincott Company 1995: 716-29.
- WISGERHOF M, BROWN RD, HOGAN MJ, CARPENTER PC, EDIS AJ. The plasma aldosterone response to angiotensin II infusion in aldosterone-producing adenoma and idiopathic hyperaldosteronism. J Clin Endocrinol Metab 1981; 52: 195-8.
- GANGULY A. Primary aldosteronism. N Engl J Med 1998; 339: 1828-34.
- IRONY I, KATER CE, BIGLIERI EG, SHACKLETON CH. Correctable subsets of primary aldosteronism. Primary adrenal hyperplasia and renin responsive adenoma. Am J Hypertens 1990; 3: 576-82.
- WILLIAMS GH, TUCK ML, ROSE LI, DLUHY RG, UNDERWOOD RH. Studies of the control of plasma aldosterone concentration in normal man. 3. Response to sodium chloride infusion. J Clin Invest 1972; 51: 2645-52.
- DLUHY RG, LIFTON RP. Glucocorticoid-remediable aldosteronism (GRA): diagnosis, variability of phenotype and regulation of potassium homeostasis. Steroids 1995; 60: 48-51.
- LITCHFIELD WR, NEW MI, COOLIDGE C, LIFTON RP, DLUHY RG. Evaluation of the dexamethasone suppression test for the diagnosis of glucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab 1997; 82: 3570-3.
- MULATERO P, VEGLIO F, PILON C, RABBIA F, ZOCCHI C, LIMONE P ET AL. Diagnosis of glucocorticoidremediable aldosteronism in primary aldosteronism: aldosterone response to dexamethasone and long polymerase chain reaction for chimeric gene. J Clin Endocrinol Metab 1998; 83: 2573-5.
- FARDELLA CE, PINTO M, MOSSO L, GÓMEZ-SÁNCHEZ C, JALIL J, MONTERO J. Genetic study of patients with dexamethasone-suppressible aldosteronism without the chimeric CYP11B1/CYP11B2 gene. J Clin Endocrinol Metab 2001; 86: 4805-7.
- MOSSO L, GÓMEZ-SÁNCHEZ CE, FOECKING MF, FARDELLA C. Serum 18-hydroxycortisol in primary aldosteronism, hypertension, and normotensives. Hypertension 2001; 38: 688-91.
- LIFTON RP, DLUHY RG, POWERS M, RICH GM, COOK S, ULICK S ET AL. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoidremediable aldosteronism and human hypertension. Nature 1992; 355: 262-5.
- JONSSON JR, KLEMM SA, TUNNY TJ, STOWASSER M, GORDON RD. A new genetic test for familial hyperaldosteronism type I aids in the detection of curable hypertension. Biochem Biophys Res Commun 1995; 207: 565-71.
- MACCONNACHIE AA, KELLY KF, MCNAMARA A, LOUGHLIN S, GATES LJ, INGLIS GC et al. Rapid diagnosis and identification of cross-over sites in patients with glucocorticoid remediable aldosteronism. J Clin Endocrinol Metab 1998; 83: 4328-31.
- CONNELL J, HAITES N, KENNEY P, LEE J. The adrenals. In: Lee JKT, Sager SS, Stanley RJ, JP H, eds. Computed body tomography with MRI correlation. Philadelphia: Lippincott-Raven Press 1998: 1171-208.
- REZNEK RH, ARMSTRONG P. The adrenal gland. Clin Endocrinol (Oxf) 1994; 40: 561-76.
- LÓPEZ JM, FARDELLA C, ARTEAGA E, MICHAUD P, RODRÍGUEZ JA, CRUZ F. Adrenal macrotumors diagnosed by computed tomography. J Endocrinol Invest 1990; 13: 581-5.
- PHILLIPS JL, WALTHER MM, PEZZULLO JC, RAYFORD W, CHOYKE PL, BERMAN AA ET AL. Predictive value of preoperative tests in discriminating bilateral adrenal hyperplasia from an aldosterone-producing adrenal adenoma. J Clin Endocrinol Metab 2000; 85: 4526-33.
- GAGNER M, POMP A, HENIFORD BT, PHARAND D, LACROIX A. Laparoscopic adrenalectomy: lessons learned from 100 consecutive procedures. Ann Surg 1997; 226: 238-46; discussion 46-7.
- SHENKER Y. Medical treatment of low-renin aldosteronism. Endocrinol Metab Clin North Am 1989; 18: 415-42.
- STOWASSER M, BACHMANN AW, HUGGARD PR, ROSSETTI TR, GORDON RD. Treatment of familial hyperaldosteronism type I: only partial suppression of adrenocorticotropin required to correct hypertension. J Clin Endocrinol Metab 2000; 85: 3313-8.