Avances en el diagnóstico de las alteraciones del eje somatotrófico que causan retardo del crecimiento
M. Isabel Hernández C.1 y Fernando Cassorla G.1
An update on the diagnosis of growth hormone axis disturbances that cause stunting
1Instituto de Investigaciones Materno Infantil. Facultad de Medicina, Universidad de Chile.
Apoyado por Proyecto Fondecyt
1060784
Correspondencia
Fernando Cassorla G
E-mail: fcassorl@med.uchile.cl
Recibido: 27 Marzo de 2009
Aceptado: 31 Abril de 2009
Human growth is a complex process regulated by several genes, most of which are unknown. Recently, our knowledge regarding the etiology of genetically determined causes of short stature has greatly increased, so molecular analysis is becoming essential for the diagnosis of growth retardation. The advances in our understanding of the molecular mechanisms involved in the function of the somatotrophic axis have resulted in a dramatic enhancement of our ability to diagnose and treat growth disorders. We hope that in the next few years improved methods for identifying specific abnormalities which cause short stature will expand our ability to diagnose other causes of growth retardation, and reduce the proportion of patients with “idiopathic” short stature.
Key words: Eje somatotrófico, talla baja, hormona del crecimiento, IGF-I.
El crecimiento somático es una de las características fundamentales de la niñez y adolescencia en el ser humano. Cabe mencionar que un niño normal crece alrededor de 25 cm durante el primer año de vida y un adolescente puede llegar a crecer alrededor de 1 cm por mes durante su estirón puberal. Este proceso es complejo y multifactorial, ya que es el producto de la interacción coordinada de diversos factores que incluyen factores genéticos, nutricionales y hormonales. En particular, uno de los elementos fundamentales de este proceso está representado por la correcta función del eje somatotrófico, ya que las alteraciones de este eje pueden modificar el patrón de crecimiento normal de un niño.
Durante los últimos años se han intentado identificar los factores genéticos que influirían en la talla final de un niño, y, específicamente, cuáles serían los genes involucrados en este proceso. En el ser humano la variación de la talla adulta es explicada en un 80% por factores genéticos, lo que confirma que es un rasgo físico altamente heredable. Los estudios de asociación genómica han evaluado más de 105 nucleótidos marcadores de polimorfismo (SNPs) a lo largo del genoma humano, en un intento de asociar determinados genotipos con talla final. Sólo 12 SNPs fueron identificados en 3 estudios epidemiológicos realizados en una amplia población de individuos normales, pero sorpresivamente ninguno de ellos correspondía a los clásicos genes asociados con el eje somatotrófico1-3.
El eje somatotrófico juega un rol clave en el crecimiento y está compuesto por una cascada hormonal que se inicia con el factor liberador de la hormona del crecimiento (GHRH) secretado por el hipotálamo. Este factor estimula la secreción pulsátil por la glándula pituitaria de la hormona de crecimiento (GH), lo que promueve el crecimiento somático y regula la composición corporal, el metabolismo óseo y la función muscular2. Algunos de los efectos de la GH son directos, mientras que otros están mediados por el factor de crecimiento insulino-símil tipo I (IGF-1) que se produce en el hígado y otros tejidos ante el estímulo con GH. La hormona IGF-1 es considerada el principal efector de la acción de GH sobre el crecimiento longitudinal, pero existen otros factores como la IGF-II y las 6 proteínas transportadoras de estos factores de crecimiento (IGFBP-1 a 6) que modulan los efectos biológicos de estas hormonas3.
Este artículo aborda las principales alteraciones del eje somatotrófico que pueden causar retardo del crecimiento en el niño. Este retardo se define como una reducción sostenida de la velocidad de crecimiento del paciente, lo que conduce a que su talla se desvíe por debajo de 2 desviaciones estándar para su patrón esperado desde el punto de vista étnico y familiar.
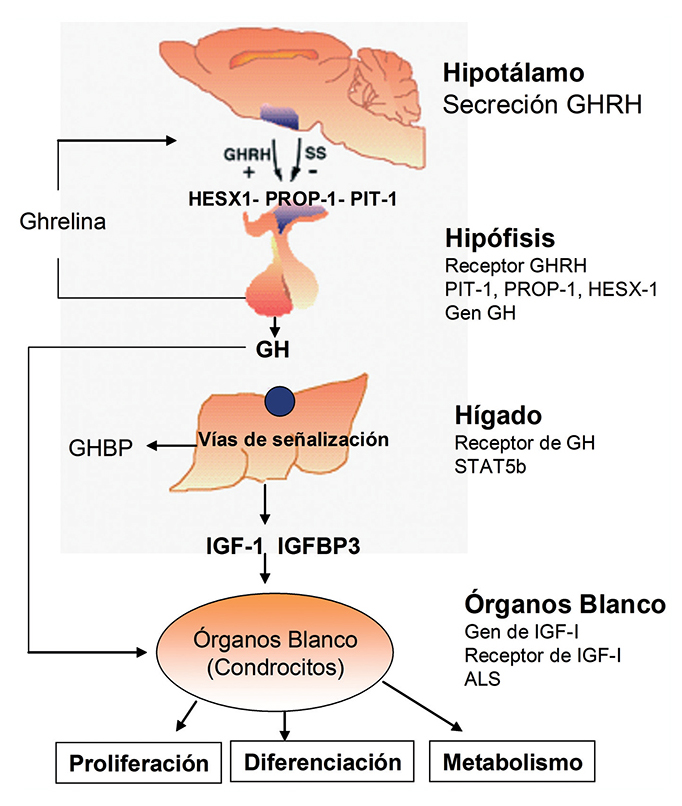
Fisiología del eje somatotrópico (Figura 1)
La regulación de la secreción de GH parece iniciarse en
la corteza cerebral que modula la secreción hipotalámica de
GHRH. Este factor hipotalámico estimula la secreción de
GH por los somatotrofos de la hipófisis, hormona que a su
vez estimula la producción de IGF-I por el hígado y otros
tejidos. Tanto la GH como la IGF-I estimulan la producción
de somatostatina por el hipotálamo, hormona que a su
vez inhibe la producción de GH. Por otra parte, la edad,
sexo, desarrollo puberal y el estado nutricional influyen
sobre la secreción de GH, así como también diversos
neurotransmisores y neuropéptidos, como la serotonina,
histamina, dopamina y GABA4. Mención especial cabe para
la Ghrelina, una hormona producida por el estómago y otros
órganos que actúa estimulando el apetito y la secreción de
GH5,6.
La secreción pulsátil de GH ha sido documentada mediante la obtención de muestras séricas frecuentes, lo que ha permitido establecer que existen picos de secreción que se presentan principalmente durante las etapas más profundas del sueño. Por esta razón no es muy útil la medición de GH en muestras aisladas7 y se debe estudiar su secreción durante una prueba de estímulo. Después de ser secretada, la GH circula unida a su proteína transportadora, mediante la formación del complejo GH-GHBP (proteína de unión a GH circulante) y posteriormente se une a sus receptores específicos en diversos tejidos8.
El receptor de GH está formado por un dominio extracelular, un dominio transmembrana y un dominio citoplasmático. La unión de GH a su receptor induce una dimerización del mismo, que estimula la fosforilación de proteínas intracelulares (principalmente JAK2), inhibición de la actividad de adenilato ciclasa y un aumento de calcio intracelular, lo que estimula a diversos activadores de la transcripción llamados STAT8.
Uno de los efectos biológicos de GH es estimular la producción de IGF-I, especialmente a nivel hepático2. Es importante mencionar que la IGF-I tiene funciones tanto endocrinas como paracrinas y autocrinas. La IGF-I está bajo el control no sólo de GH, sino que también de otros factores, en especial nutricionales. En el torrente sistémico, las IGFs circulan unidas a sus proteínas transportadoras (IGFBPs), las que permiten liberar IGF en forma específica en los diversos tejidos. Dentro de estas proteínas transportadoras, la IGFBP-3 es especialmente dependiente de GH9. La unión de IGF-I a IGFBP-3 y a la subunidad ácido-lábil (ALS) forma un complejo ternario que proporciona estabilidad a la IGF-I circulante. Finalmente, la IGF-I libre se une a su receptor (IGF-IR) ubicado en distintos órganos, lo que estimula el crecimiento somático, especialmente debido a sus efectos sobre el cartílago de crecimiento5.
Alteraciones del eje somatotrófico que causan retardo del crecimiento
Desde la primera descripción de pacientes con insensibilidad a GH, publicada por Laron en el año 196610, y que posteriormente se aclaró que es causada por mutaciones en el receptor de GH, mucho se ha avanzado en el diagnóstico de las diferentes causas moleculares de talla baja. Durante los últimos años se han identificado diversos pacientes con patologías desconocidas previamente que pueden causar alteraciones del eje somatotrófico y retardo del crecimiento.
1. Alteraciones a nivel hipotalámico
Alteraciones en la secreción de GHRH: En algunos pacientes con deficiencia de GH se pueden documentar alteraciones en la secreción del GHRH11. Estos pacientes no tienen deficiencia de GH, ya que producen GH en forma normal en respuesta al estímulo externo con el factor liberador, por lo que el defecto se ubica en el hipotálamo y no en la hipófisis. En estos casos el tratamiento con factor liberador puede producir mejoría en la velocidad de crecimiento.
Mutaciones del receptor de GHRH: Las mutaciones en el gen del receptor de GHRH12 pueden causar un profundo retraso de crecimiento. Estas mutaciones han sido descritas en grupos consanguíneos de diferentes partes del mundo, especialmente en zonas aisladas de Pakistán13 y Brasil14. La característica de esta alteración radica en que estos pacientes no aumentan sus niveles de GH al ser estimulados con GHRH. Pese a que el hipotálamo y la hipófisis son capaces de producir las hormonas GHRH y GH en forma normal, respectivamente, al existir la alteración a nivel del receptor de GHRH este factor liberador no induce la producción de GH. Estos pacientes tienen una estatura final muy reducida, pero no tienen alteración en la secreción de otras hormonas hipofisiarias. El tratamiento con GH en ellos mejora su velocidad de crecimiento.
Mutaciones del receptor de Ghrelina: La Ghrelina es un péptido orexígeno producido predominantemente en el estómago, que estimula la secreción de GH por la glándula pituitaria. Se han descrito mutaciones en el receptor de Ghrelina en algunos pacientes con talla baja15. Ellos exhiben niveles circulantes reducidos de GH, con una pobre respuesta a las pruebas de estímulo de secreción de GH y tienen además niveles bajos de IGF-I e IGFBP-3.
Tabla 1. Alteraciones del eje somatotrófico que causan retardo del crecimiento

2. Alteraciones hipofisiarias
Los somatotrófos se ubican en la adenohipófisis, cons– tituyendo aproximadamente el 50% de las células de esta glándula. Para que se produzca la diferenciación adecuada de los somatotrófos desde sus células progenitoras durante la embriogénesis, se requiere la participación coordinada de diversos factores de transcripción, entre los cuales se encuentran el PIT-1 y PROP-1. Estos factores no sólo regulan la diferenciación y proliferación de los somatotrófos, sino que de otras células de la adenohipófisis como los lactotrofos, gonadotrofos, tirotrofos y corticotrofos.
Alteraciones de los factores de transcripción hipfisiarios: Se han descrito diversas mutaciones en los genes involucrados en la formación de la hipófisis, específicamente en PIT1, PROP1, HESX1, LHX3 y LHX4, que se pueden presentar clínicamente como un déficit en la síntesis y secreción de GH16. Mutaciones en los genes PIT1 y PROP-1 pueden ser responsables de un retraso profundo en el crecimiento, causado por la deficiencia combinada de GH, TSH, prolactina y ocasionalmente de gonadotropinas y ACTH, en el caso de PROP-116. Cabe mencionar que la prevalencia de mutaciones en PROP1 en pacientes con hipopituitarismo congénito es relativamente alta.
Otras mutaciones como las del HESX1, que se han asociado con displasia septo-óptica, parecen ocurrir con menos frecuencia17. Este cuadro clínico está caracterizado por alteraciones neurológicas, específicamente con hipoplasia del septum pellucidum, agenesia del cuerpo calloso y atrofia del nervio óptico, que se puede manifestar con ceguera y alteraciones endocrinas como deficiencia de GH y diabetes insípida. Las mutaciones del gen LHX3 son relativamente infrecuentes y habitualmente causan deficiencias de GH, TSH, LH, FSH y PRL que pueden estar asociadas a limitaciones en la rotación del cuello18. Los pacientes con mutaciones del gen LHX4 pueden exhibir una forma de hipopituitarismo congénito asociado con anormalidades en el desarrollo del encéfalo como la malformación de Chiari19.
Mutaciones del gen de GH: La GH es una hormona polipeptídica de 191 aminoácidos, que está codificada por un gen ubicado en el brazo largo del cromosoma 17. Existen 5 genes que codifican distintas especies moleculares de GH, lo que significa que la GH se encuentra en la circulación en diversas formas que tienen una alta homología estructural20. La GH1 es la forma molecular más importante que se expresa en la hipófisis y da origen a la mayor parte de la hormona de crecimiento circulante. Los otros 4 genes para GH se expresan en diversos tejidos como la placenta.
Se han descrito diversos defectos moleculares, entre ellos mutaciones y deleciones en el gen o en el promotor del gen de GH121,22. Clínicamente la mayoría de estos pacientes exhiben una pronunciada reducción de su velocidad de crecimiento. También se ha sugerido que existirían casos de GH con menor potencia biológica, pero estas alteraciones serían poco frecuentes6,16.
Las alteraciones estructurales en el gen de GH causan una deficiencia aislada de GH, que se puede manifestar con distintos patrones de herencia:
I-A Autosómica recesiva: Causada por una deleción del gen de GH, por lo que no existen niveles circulantes detectables de GH. Representa la forma más severa de deficiencia de GH. La respuesta a GH es variable, ya que estos pacientes pueden desarrollar anticuerpos durante el tratamiento con GH exógena.
I-B Autosómica recesiva: Causada por mutaciones en el gen de GH, por lo que existen concentraciones séricas disminuídas, pero medibles, de GH. Estos pacientes habitualmente responden en forma apropiada al tratamiento con GH.
II Autosómica dominante: Causada por mutaciones en el gen de GH, por lo que el cuadro clínico es muy similar al tipo I-B, pero con un patrón de herencia distinto, ya que estos pacientes tienen a un progenitor con el mismo cuadro clínico.
III Ligada al X: Su modo de transmisión está ligada al cromosoma X y su expresión clínica es algo variable, aunque similar al tipo I-B. Algunos de estos pacientes pueden manifestar un cuadro de agamaglobulinemia asociado.
3. Insensibilidad a GH
Existen una variedad de alteraciones moleculares en el receptor de GH y en la señalización intracelular para GH que conforman las diferentes formas del síndrome global de insensibilidad a GH. Este cuadro puede clasificarse en insensibilidad primaria o secundaria a GH23. La insen– sibilidad primaria puede estar causada por alteraciones estructurales en el receptor de GH o en los sustratos intracelulares para la transmisión de la señal para GH como STAT5b. La insensibilidad secundaria a GH puede deberse a la presencia de desnutrición, enfermedades sistémicas graves, en especial las hepáticas, o de anticuerpos contra el receptor de GH, que pueden afectar la transmisión de señal para esta hormona en diversos tejidos.
Alteraciones primarias del receptor de GH: Se han descrito una variedad de mutaciones en la porción extracelular, transmembranosa e intracelular del receptor de GH24.El cuadro se transmite en forma autosómica recesiva, por lo que se observa en núcleos familiares consanguíneos descritos en poblaciones del Medio Oriente y del sur de Ecuador. En nuestro país hemos publicado algunos casos de esta patología25. Estos pacientes presentan un importante retraso del crecimiento, predominantemente post natal, que se puede asociar con un fenotipo caracterizado por frente prominente, facies infantil, puente nasal hipoplásico, voz aguda, además de obesidad e hipogenitalismo. Debido a la alteración del receptor de GH, estos pacientes exhiben concentraciones elevadas de GH y reducidas de IGF-I e IGFBP-326, y no responden al tratamiento con dosis habituales de GH exógena, por lo que deben ser tratados con dosis altas de GH, o preferentemente con IGF-I.
Es importante destacar que existe un número creciente de pacientes con insensibilidad parcial a GH causadas por mutaciones sutiles en el receptor de GH, que pueden haber sido erróneamente catalogados como portadores de una “talla baja idiopática”27. Por lo tanto, el espectro de esta patología se ha ampliado considerablemente al estudiar la estructura genómica del receptor de GH en pacientes con retardo del crecimiento asociados a niveles circulantes relativamente elevados de GH y disminuidos de IGF-I. Este estudio permite además seleccionar la terapia hormonal más apropiada para estos pacientes.
Mutaciones de STAT5b: Otra forma primaria de insensibilidad a GH está representada por mutaciones en el gen de STAT5b, inicialmente descrita por Kofoed en el año 2003 en una paciente de 16 años con una talla -7,5 DS y concentraciones reducidas de IGF-I, IGFBP-3 y ALS, a pesar de exhibir una estructura normal del gen del receptor de GH. En esta paciente se encontró una mutación homozigota en una región altamente conservada del gen para STAT5b28. Posteriormente, se han descrito otras pacientes con mutaciones en este importante sustrato intracelular, la mayoría mujeres, caracterizadas por presentar un cuadro asociado de inmunodeficiencia severa e hiperprolactinemia. Cabe mencionar que hasta la fecha no se han descrito mutaciones en los genes para otros sustratos intracelulares como JAK2 o MAPK que produzcan un cuadro de insensibilidad a GH.
4. Mutaciones de IGF-I y del complejo ternario
Los factores de crecimiento insulino símiles, son péptidos con estructura semejante a la insulina. Su principal función es ser mediadores de la acción de GH en los órganos blanco, jugando un rol clave en el crecimiento pre y postnatal. El crecimiento fetal es muy dependiente de IGFII, mientras que durante la niñez y adolescencia la IGF-I es el más potente estimulador del crecimiento y diferenciación celular. A nivel de la placa epifisiaria, la IGF-I estimula la diferenciación y multiplicación de los condrocitos, lo que promueve el crecimiento de los huesos largos. Este efecto de IGF-I es parcialmente estimulado por GH, lo que hace que la medición de IGF-I sea muy importante para establecer el diagnóstico de deficiencia de GH.
Cabe mencionar que los niveles de IGF-I aumentan en forma muy significativa desde el nacimiento hasta la adolescencia y presentan un marcado dimorfismo sexual, con valores más elevados en las mujeres29. Además las concentraciones circulantes de IGF-I son muy dependientes de un satisfactorio estado nutricional y una adecuada función hepática.
Se han descrito diversos defectos en la producción, transporte y acción de IGF-I, tales como deleciones del gen de IGF-I, mutaciones del complejo ternario y del receptor de IGF-I.
Mutaciones en el gen de IGF-I: En 1996 Woods y cols, describieron al primer paciente con una deleción homozigota de los exones 4 y 5 del gen de IGF-I. Clínicamente este sujeto había evidenciado una dismorfia caracterizada por ptosis y micrognatia, asociada a retardo del crecimiento intrauterino y del crecimiento postnatal, microcefalia, retardo mental y sordera sensorio-neural30. Este paciente exhibía niveles indetectables de IGF-I sérica, asociado a concentraciones elevadas de GH, pero su edad ósea no estaba muy atrasada. El paciente fue tratado por aproximadamente dos años con GH sin cambios en su velocidad de crecimiento. Posteriormente, se han descrito otros casos de mutaciones puntuales en el gen de IGF-I, algunos en individuos adultos que han manifestado características clínicas similares al caso de Woods31,32.
Mutaciones en la unidad ácido lábil (ALS) del complejo ternario: Recientemente, se han descrito algunos casos de mutaciones en el gen de ALS, componente fundamental del complejo ternario, compuesto por IGF-I, IGFBP-3 y ALS. Este cuadro está caracterizado por un leve retardo del crecimiento con retraso del desarrollo puberal, y está asociado a una importante disminución en las concentraciones circulantes de IGF-I e IGFBP-333,34. Estos casos proporcionan importante información sobre el rol del complejo ternario sobre el crecimiento y desarrollo puberal en el ser humano. Queda por definir la prevalencia de este cuadro en pacientes con estas características clínicas, pero que son relativamente sutiles y que por lo tanto, no han sido estudiados en forma sistemática.
Mutaciones del receptor de IGF-I: Recientemente, se han descrito algunos casos de mutaciones en el receptor de IGF-I en el ser humano, que resultan en una alteración de la transmisión de la señal intracelular para esta importante hormona. Las mutaciones homozigotas del receptor de IGF-I en modelos murinos “knock-out” producen un significativo retardo del crecimiento intrauterino y conducen a una elevada mortalidad neonatal, mientras que las mutaciones heterozigotas no parecen producir patología. La primera descripción de mutaciones en el receptor de IGF-I en el ser humano fue realizado por Abuzzahab et al, el año 200335. Los casos descritos hasta ahora se caracterizan por retardo del crecimiento intrauterino y del crecimiento postnatal, y leve retardo mental. Las concentraciones séricas de IGF-I pueden ser normales o elevadas, pero la unión de IGF-I a su receptor está disminuida por una alteración estructural del receptor. El manejo clínico de estos pacientes es difícil, aunque algunos pueden responder al tratamiento con dosis elevadas de GH o IGF-I.
Conclusiones
En esta revisión hemos presentado los avances más recientes en las alteraciones del eje somatotrófico que pueden causar retardo del crecimiento en el niño. Lejos están los tiempos en que el endocrinólogo podía evaluar la función del eje somatotrófico sólo con una prueba de estímulo para determinar la secreción de GH. Actualmente, es necesario investigar con mucha acuciosidad los diferentes componentes de este eje, que incluyen los niveles circulantes de IGF-I e IGFBP-3, con lo que se puede dirigir el estudio de determinados pacientes hacia posibles mutaciones en los diversos genes involucrados en el proceso del crecimiento infantil.
En base a la relación entre la presentación clínica del paciente y las concentraciones de GH, IGF-I e IGFBP-3, se puede orientar el diagnóstico hacia disfunciones más sutiles del eje somatotrófico, entre las que se encuentran alteraciones en la secreción y acción de los péptidos liberadores de GH, en la expresión de factores hipofisiarios de la transcripción, en la estructura de los genes para GH, IGF-I y ALS y en la transmisión de señal intracelular para GH e IGF-I. A pesar del vasto avance que se ha logrado en este campo hasta la fecha, es indudable que en el futuro próximo se descubrirán nuevos mecanismos patogénicos que causan retardo del crecimiento en el niño. Esto permitirá diagnosticar otras causas de talla baja, aún clasificadas como idiopáticas y ofrecer un mejor tratamiento para esta patología tan prevalerte en el consultorio de endocrinología infantil.
Referencias
- Hernández MI, Cohen P. 2008. Surprising new height regulating genes: beyond growth hormone and IGF-I. Pediatr Res 64: 461
- Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, et al. 2008. Identification of ten loci associated with height highlights new biological pathways in human growth.Nat Genet 40: 584-91.
- Etherton TD. 2004. Somatotropic function: the somatomedin hypothesis revisited.J Anim Sci.82 E-Suppl: E239-244.
- Kaplan SA, Cohen P. 2007. The somatomedin hypothesis 2007: 50 years later. J Clin Endocrinol Metab 92: 4529-35.
- Walenkamp MJ, Wit JM. 2007. Genetic disorders in the GH IGF-I axis in mouse and man. Eur J Endocrinol 157; Suppl 1: S15-26
- Rosenfeld R, Cohen P. 2008. Disorders of growth hormone/ insulinlike growth factor secretion and action. En Sperling (ed) Pediatric Endocrinology. Philadelphia, US. Saunders-Elsevier 254-334.
- Veldhuis JD, Carlson ML, Johnson ML. 1987. The pituitary gland secretes in bursts: appraising the nature of glandular secretory impulses by simultaneous multiple-parameter deconvolution of plasma hormone concentrations. Proc Natl Acad Sci USA 84: 7686-7690.
- Bougnères P, Goffin V. 2007. The growth hormone receptor in growth. Endocrinol Metab Clin North Am 36: 1-16.
- Rosenfeld RG, Lamson G, Pham H, Oh Y, Conover C, De Leon DD, et al. 1990; Insulin like growth factor-binding proteins. Recent Prog Horm Res 46: 99-159.
- Laron Z, Pertzelan A, Mannheimer S. 1966. Genetic pituitary dwarfism with high serum concentration of growth hormone. A new inborn error of metabolism? Isr J Med Sci 2: 153-155.
- Lin-Su K, Wajnrajch MP. 2002. Growth Hormone Releasing Hormone (GHRH) and the GHRH Receptor. Rev Endocr Metab Disord 3: 313-323.
- Wajnrajch MP, Gertner JM, Harbison MD, Chua SC Jr, Leibel RL. 1996. Nonsense mutation in the human growth hormone-releasing hormone receptor causes growth failure analogous to the little (lit) mouse. Nat Genet 12: 88-90.
- Baumann G, Maheshwari H. 2007. The Dwarfs of Sindh: severe growth hormone (GH) deficiency caused by a mutation in the GHreleasing hormone receptor gene. Acta Paediatr Suppl 423: 33-38.
- Salvatori R, Hayashida CY, Aguiar-Oliveira MH, Phillips JA 3rd, Souza AH, et al. 1999. Familial dwarfism due to a novel mutation of the growth hormone-releasing hormone receptor gene. J Clin Endocrinol Metab 84: 917-923.
- Pantel J, Legendre M, Cabrol S, Hilal L, Hajaji Y, et al. 2006. Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J Clin Invest 116: 760-768.
- Mullis PE. 2005. Genetic control of growth. Eur J Endocrinol 152: 11-31.
- McNay DE, Turton JP, Kelberman D, Woods KS, Brauner R, et al. 2007. HESX1 mutations are an uncommon cause of septo optic dysplasia and hypopituitarism. J Clin Endocrinol Metab 92: 691-697.
- Pfaeffle RW, Savage JJ, Hunter CS, Palme C, Ahlmann M, et al. 2007. Four novel mutations of the LHX3 gene cause combined pituitary hormone deficiencies with or without limited neck rotation. J Clin Endocrinol Metab 92: 1909-1919.
- Castinetti F, Saveanu A, Reynaud R, Quentien MH, Buffin A, Brauner R, et al. 2008. A novel dysfunctional LHX4 mutation with high phenotypical variability in patients with hypopituitarism. J Clin Endocrinol Metab 93: 2790-2799.
- Miller WL, Eberhardt NL. 1983. Structure and evolution of the growth hormone gene family. Endocr Rev 4: 97-130.
- Takahashi Y, Kaji H, Okimura Y, Goji K, Abe H, Chihara K. 1983. Brief report: short stature caused by a mutant growth hormone. N Engl J Med 334: 432-436.
- Takahashi Y, Shirono H, Arisaka O, Takahashi K, Yagi T, et al. 1997. Biologically inactive growth hormone caused by an amino acid substitution. J Clin Invest 100: 1159-1165.
- Kaplan SA, Cohen P. The somatomedin hypothesis 2007: 50 years later 2007 J Clin Endocrinol Metab 92: 4529-4535.
- Laron Z. 2002. Growth hormone insensitivity (Laron syndrome). Rev Endocr Metab Disord 3: 347-355.
- Espinosa C, Sjoberg M, Salazar T, Rodríguez A, Cassorla FG, et al. 2008. E180splice mutation in the growth hormone receptor gene in a Chilean family with growth hormone insensitivity: a probable common Mediterranean ancestor. J Pediatr Endocrinol Metab 21: 1119-1127.
- Rosenbloom AL, Guevara Aguirre J, Rosenfeld RG, Fielder PJ. 1990. The little women of Loja--growth hormone-receptor deficiency in an inbred population of southern Ecuador N Engl J Med 323: 1367-1374.
- Bonioli E, Tarò M, Rosa CL, Citana A, Bertorelli R, et al.2005 Heterozygous mutations of growth hormone receptor gene in children with idiopathic short stature. Growth Horm IGF Res; 15: 405-410.
- Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, et al. 2003. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med 349: 1139-1147.
- Cara JF, Rosenfield RL, Furlanetto RW. 1987. A longitudinal study of the relationship of plasma somatomedin-C concentration to the pubertal growth spurt. Am J Dis Child; 141: 562-564.
- . Woods KA, Camacho-Hübner C, Savage MO, Clark AJ. 1996. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene N Engl J Med 335: 1363-1367.
- Bonapace G, Concolino D, Formicola S, Strisciuglio P 2003 A novel mutation in a patient with insulin-like growth factor 1 (IGF1) deficiency. J Med Genet 40: 913-917.
- Walenkamp MJ, Karperien M, Pereira AM, Hilhorst-Hofstee Y, van Doorn J, et al. 2005. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J Clin Endocrinol Metab 90: 2855-2864.
- . Domené HM, Bengolea SV, Martínez AS, Ropelato MG, Pennisi P, et al. 2004. Deficiency of the circulating insulin-like growth factor system associated with inactivation of the acid-labile subunit gene N Engl J Med 350: 570-577.
- Hwa V, Haeusler G, Pratt KL, Little BM, Frisch H, et al. 2006. Total absence of functional acid labile subunit, resulting in severe insulin-like growth factor deficiency and moderate growth failure J Clin Endocrinol Metab 91: 1826-1831.
- Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, et al. 2003. Intrauterine Growth Retardation (IUGR) Study Group. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med 349: 2211-2222.