Infundibuloneurohipofisitis. Caso clínico y revisión
de la literatura
Patricio Salman M.1
Infundibuloneurohypophysitis: Clinical report and literature review
1Sección Endocrinología, Departamento de Medicina Interna de la Facultad de Medicina, Universidad de Concepción. Concepción, Chile.
Sin financiamiento. Sin conflicto de interés.
Correspondencia:
Patricio Salman Mardones
Profesor asistente de la Facultad de Medicina de la Universidad de Concepción.
Víctor Lamas 1290, Barrio Universitario, Concepción.>
Teléfono: (041) 2204921.>
psalman@udec.cl
Recibido: 17/04/2017
Aceptado: 12/06/2017
Infundibuloneurohypophysitis is a rare condition, which is part of the group of hypophysitis, of relatively recent description (1993). The main clinical manifestation is diabetes insipidus, whose natural evolution is towards chronicity. The differential diagnosis with other thickening of the hypophysial stem is very important, where the clinic, imaging, laboratory and eventually biopsy are a main support for a correct diagnosis. We present a clinical case that shows the usual picture of infundibuloneurohypophysitis, and illustrates the imaging evolution in a female patient, with diabetes insipidus as the main clinical manifestation.
Key words: Hypophysitis, Infundibuloneurohypophysitis, Diabetes Insipidus.
1Sección Endocrinología, Departamento de Medicina Interna de la Facultad de Medicina, Universidad de Concepción. Concepción, Chile.
La hipofisitis es un cuadro caracterizado por la infiltración lifocítica de la glándula pituitaria. Dentro de su clasificación encontramos la infundibuloneurohipofisitis que afecta principalmente al tallo hipofisiario, infundíbulo y neurohipófisis. Su principal cuadro clínico es la diabetes insípida central, que lo diferencia habitualmente del compromiso global de la pituitaria o panhipofisitis. Tiene generalmente un curso crónico que puede ser adecuadamente manejado con desmopresina. A partir de un caso clínico presentamos el curso natural de esta enfermedad e ilustramos los cambios anatómicos clásicos en la región selar a través de la resonancia magnética.
Caso clínico
Paciente sexo femenino, de 31 años, con historia de embarazo a fines de 2012, donde comienza con cuadro de polidipsia inicialmente sin poliuria. A medida que el embarazo avanzó se agregó poliuria manifiesta. No se hizo mayores estudios en ese embarazo y su hijo nació en Estados Unidos, aparentemente sin complicaciones. Del punto de vista obstétrico hubo una importante hemorragia uterina post parto inmediato que se resolvió médicamente.
Dentro de sus antecedentes, paciente sana previa, en su familia sólo destaca una hermana con miastenia gravis, en amenorrea desde el parto, no fumadora, no bebedora de alcohol.
Posterior al embarazo se mantuvo la polidipsia y poliuria llegando a una diuresis de más de 10 litros al día, con nicturia de 4 a 5 veces cada noche. Consultó por la sintomatología descrita y un médico no especialista le diagnosticó diabetes insípida y le indicó hidroclorotiazida que al cabo de 2 meses de no haber respuesta se cambió por DDAVP 2 puff al día notando gran mejoría en particular en la poliuria. Importante mencionar que no se hizo en ese momento test de deprivación acuosa.
Posteriormente, consultó con especialista endocrinólogo donde se le solicitó exámenes que destacaba: glicemia 97 mg/dl, creatininemia 0,71 mg/dl, sodio 145 mEq/l, potasio 3,6 mEq/l, cloro 105 mEq/l, FSH 3,8 MUI/ml, Estradiol 12 pg/ml, prolactina 20,6 ng/ml, TSH 4,69 UI/ml, T4 libre 1,0 ng/dl, IGF-1 30,8 ng/ml (130-354), cortisol 151 ng/ml (40-230). Resonancia magnética (RM) de la región selar informó “silla turca normal, engrosamiento nodular del tercio medio distal del tallo hipofisiario y no se identifica señal de la neurohipófisis” (Figura 1). Se ajustó DDAVP a 2 puff en la mañana y 1 puff en la tarde, logrando ya normalizar la diuresis diurna y no tener nicturia. Se solicitó control RM en forma precoz para evaluar evolución de engrosamiento del tallo y segunda resonancia informó “no se identifica señal de neurohipófisis, evolución regresiva del engrosamiento del tallo hipofisiario respecto a RM previa” (Figura 2). Se establece diagnóstico de Infundibuloneurohipofisitis, Diabetes Insípida central y amenorrea secundaria por hipogonadismo hipogonadotrópico. La paciente evoluciona en forma adecuada, asintomática, con completo control de la diuresis, y dado el compromiso gonadal y la edad se inició terapia reemplazo hormonal. Siguiente RM evidenció normalización del grosor del tallo hipofisiario, sin evidenciar la neurohipófisis.
Cabe mencionar que en una ocasión la paciente viajó un fin de semana donde se le olvidó usar el DDAVP y presentó nuevamente importante poliuria y nicturia que se normalizó una vez reiniciado el DDAVP volviendo de su viaje. Este hecho, establece con seguridad que la paciente tendrá una diabetes insípida permanente y el uso de la desmopresina será crónico.
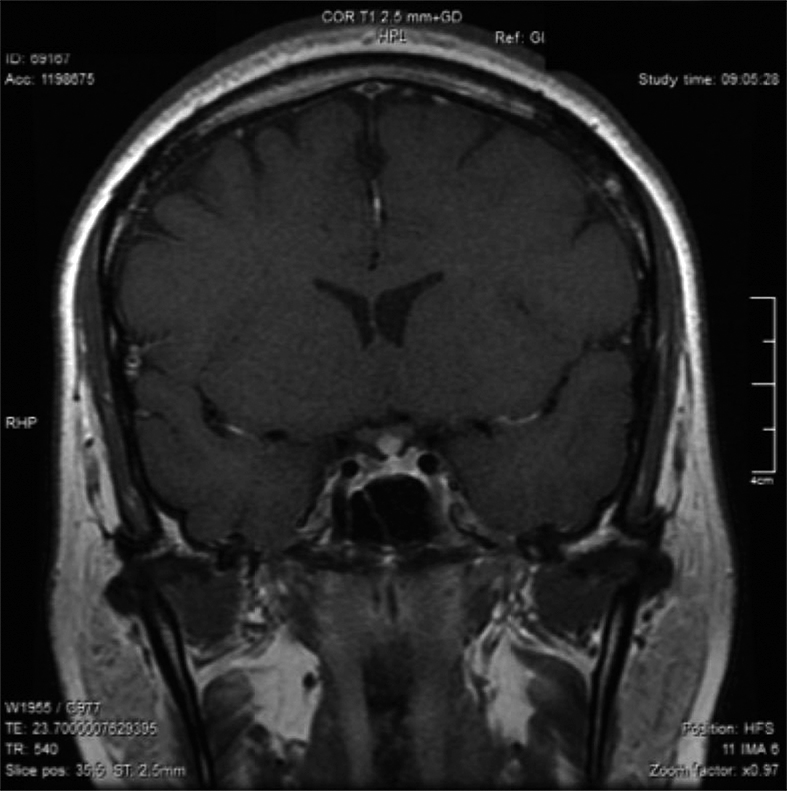 Figura 1. Engrosamiento nodular del tercio medio distal del tallo hipofisiario.
Figura 1. Engrosamiento nodular del tercio medio distal del tallo hipofisiario. 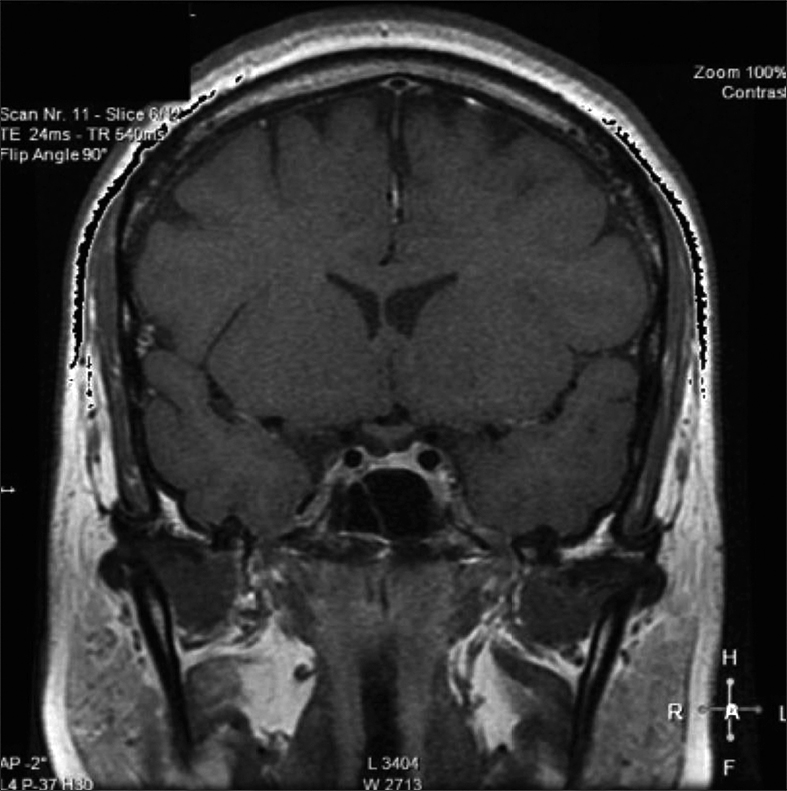 Figura 2. Evolución regresiva del engrosamiento del tallo hipofisiario.
Figura 2. Evolución regresiva del engrosamiento del tallo hipofisiario.
Discusión
La infundibuloneurohipofisitis (INH) es un cuadro inflamatorio poco habitual que se ha postulado ser de origen autoinmune. Se enmarca dentro un concepto más amplio que es la hipofisitis. Esta última se clasifica de acuerdo a la zona anatómica comprometida por el proceso inflamatorio y que tiene habitualmente correlación con las alteraciones hormonales de la zona afectada.
La hipofisitis se clasifica en: Adenohipofisitis (AH), Infundibuloneurohipofisitis (INH) y Panhipofisitis (PH)1. Del punto de vista de los hallazgos histopatológicos la hipofisitis se clasifica en: linfocítica, granulomatosa, xantomatosa, relacionada a IgG4, necrotizante y formas mixtas1. Este cuadro fue reportado por primera vez en 1962 por Goudie y Pinkerson en una mujer que post parto presentó hipotiroidismo y amenorrea, que posteriormente falleció de insuficiencia suprarrenal aguda2. La prevalencia de la hipofisitis linfocítica es alrededor de 0,24 a 0,88%1, su incidencia anual es 1 en 9 millones, aunque probablemente es subestimado3. Según histopatología la forma más frecuente es la linfocítica (71,8%) seguido de la granulomatosa (18,6%)4. Generalmente, ocurre en la cuarta década de la vida, siendo rara en niños y en ancianos5. La AH linfocítica es 4-6 veces más frecuente en mujeres, especialmente en embarazo y post parto. La INH afecta a hombre y mujeres por igual5,6. El cuadro clínico de la hipofisitis es variable y pueden ocurrir cualquiera de estas 4 presentaciones: a) Efecto de masa (cefalea, déficit visual con compromiso de quiasma, III-IV o VI pares craneales, diplopia); b) Hipopituitarismo (habitualmente con deficiencia en el siguiente orden: ACTH, luego TSH, luego LH/FSH, luego prolactina, luego GH); c) Diabetes insípida central (en el 14 a 20% de los casos); d) Hiperprolactinemia1. Esto contrasta con la INH donde la diabetes insípida es la presentación clínica cardinal, tal como ocurrió con nuestra paciente.
Respecto a la INH fue reportada por primera vez en 1970 y luego en 1993 por Saito et al e Imura et al respectivamente a raíz de diabetes insípida idiopática, donde el diagnóstico de infundibuloneurohipofisitis fue realizado post mortem7,8; en estos casos la infiltración linfocítica fue limitada sólo al infundíbulo, tallo y neurohipófisis. Condiciones autoinmunes concomitantes están asociados a INH en un 20-25% de los casos, tales como tiroiditis de Hashimoto, Síndrome Pluriglandular Autoinmune, Enfermedad de Basedow Graves, Lupus Eritematoso Sistémico, Síndrome Sjögren, Beçhet, Diabetes tipo 1, entre otros. La INH ocurre habitualmente más tardiamente (más de 50 años) que la AH y la PH, a diferencia de nuestro caso donde fue una paciente joven de 31 años.
Como se mencionó, la forma de presentación de la INH es la diabetes insípida de origen central, y en general no hay compromiso de la adenohipófisis, aunque hiposecreción de GH y LH/FSH están descritas. Por otro lado, si bien la INH ha sido reportada en el contexto de embarazo, la presentación en el período post parto ha sido infrecuentemente descrita9. En el caso de nuestra paciente la sintomatología comenzó durante su embarazo y se perpetuó post parto, y si bien hubiese sido deseable, cuando se controló la primera vez, un test de deprivación acuosa, éste ya no fue necesario cuando se empezó a controlar en endocrinología debido que el cuadro clínico de diabetes insípida era claro, mejoró notablemente con DDAVP, y la RM evidenció un engrosamiento del tallo hipofisiario y la ausencia de la señal de la neurohipófisis. Nuestra paciente también presentó amenorrea que se confirmó posteriormente como un hipogonadismo hipogonadotrópico, y si bien no se puede descartar que haya habido algún compromiso de adenohipofisitis, es planetable también que haya sido producto de la importante hemorragia post parto inmediato que la paciente presentó.
Dentro del correcto diagnóstico de la INH está la neuroimagen, donde la RM es el método de elección. Los hallazgos comúnmente encontrados son el engrosamiento del infundíbulo y del tallo hipofisiario (se considera engrosamiento si es mayor a 4 mm) y la pérdida de señal de la neurohipófisis (hay que considerar que el 10% de la población normal no la presenta, especialmente en ancianos). Esta asociación, ausencia de señal de neurohipófisis y engrosamiento del tallo hipofisiario, son altamente sugerentes de INH. En nuestra paciente se encontraron ambos fenómenos, y considerando la regresión del engrosamiento del tallo hipofisiario lo hacía compatible con INH. La adenohipófisis en la RM es usualmente normal en tamaño e intesidad a la señal. Siendo el engrosamiento del tallo hipofisiario el hallazgo cardinal a la imagenología, inicialmente se le denominaba “Stalkitis” a esta entidad10; sin embargo, la denominación INH es la más idónea debido a que anatomopatológicamente el compromiso es del infundíbulo, tallo y neurohipófisis. Respecto al engrosamiento del tallo hipofisiario en muy importante considerar su diagnóstico diferencial9. Una adecuada historia clínica, manifestaciones clínicas, imagenología y en ocasiones biopsia darán el diagnóstico adecuado. En la Tabla 1 se enumeran las etiologías de engrosamientos del tallo hipofisiario aisladas.
Tabla 1. Etilogías de engrosamiento del tallo hipofisiario
| Autoinmune | - Infundibuloneurohipofisitis linfocítica |
| Neoplásicas | - Germinoma |
| Inflamatorias/granulomatosas | - Histiocitosis |
| Infecciosas | - Tuberculosis |
| Medicamentos | - Ipilimumab |
Respecto al rol de la biopsia en la INH el clínico debe poner en una balanza el riesgo de la biopsia pituitaria en el contexto de los hallazgos clínicos y radiológicos. En la actualidad el diagnóstico de INH puede ser establecido por el cuadro clínico y la RM característica. No obstante, es muy relevante el seguimiento precoz con neuroimagen para constatar la regresión del engrosamiento del tallo hipofisiario o que no haya aumentado de tamaño. Por lo tanto, si bien no es de regla la biopsia, debe considerarse fuertemente su realización ante la duda diagnóstica, en particular ante la sospecha de causas neoplásicas como germinoma o linfoma9.
El manejo de la INH es el manejo de la diabetes insípida central, con DDAVP. El rol de los corticoides es controversial dado los resultados contradictorios en la literatura. La gran mayoría de las INH son autolimitados y no es necesario el uso de corticoides. Se plantea la cirugía sólo en caso de efecto de masa con compresión de nervios craneanos, en particular si no hay respuesta a corticoides si se utilizaron. La diabetes insípida puede ser un fenómeno transitorio, pero la literatura muestra que generalmente es permanente y el uso del DDVAP se vuelve crónico1,9,11.
En resumen, presentamos el caso ilustrativo de INH con la imagenología clásica, y su total regresión del engrosamiento del tallo hipofisiario y la clínica habitual que es la diabetes insípida.
Referencias bibliográficas
- Fukoka H. Hypophysitis. 2015. Endocrinol Metab Clin N Am; 44: 143-9.
- Goudie RB, Pinkerton PH. 1962. Anterior hypophysitis and Hashimoto´s disease in a young woman. J Pathol Bacteriol 83: 584-5.
- Buxton N, Robertson I. 2001. Lymphocytic and granulocytic hypophysitis: a single centre experience. Br J Neurosurg 15: 242-5.
- Caturegli P, Iwama S. 2013. From Japan with love: another tessera in the hypophysitis mosaic. J Clin Endocrinol Metab 98: 1865-8.
- Caturegli P, Lupi I, Landek-Salgado M, et al. 2008. Pituitary autoimmunity: 30 years later. Autoimmun Rev 7: 631-7.
- Falorni A, Minarelli V, Bartoloni E, et al. 2014. Diagnosis and classification of autoimmune hypophysitis. Autoimmun Rev 13: 412-6.
- Saito T, Yoshida S, Nakao K, et al. 1970. Chronic hypernatremia associated with inflammation of the neurohypophysis. J Clin Endocrinol Metab 31: 391-6.
- Imura H, Nakao K, Shimatsu A, et al. 1993. Lymphocytic infundibuloneurohypophysitis as a cause of central diabetes insipidus. N Engl J Med 329: 683-9.
- Philip CJ, Luen SC, Amir HH, et al. 2015. Lymphocytic infundibulo-neurophypophysitis: a clinical overview. Endocrine (50); 3: 531-6.
- Legget D, Hill P, Anderson R. 1999. “Stalkitis” in a pregnant 32 year old woman: A rare cause of diabetes insipidus. Australasian Radiology 43: 104-7.
- Abe T. 2008. Lymphocytic infundibulo-neurohypophysitis and infundibulo-panhypophysitis regarded as lymphocytic hypophysitis variant. Brian Tumor Pathol 25: 59-66.