Efecto de Estradiol y Testosterona sobre la activación de la vía JAK2/STAT5 inducida por hormona de crecimiento
Paula Ocaranza, Germán Íñiguez, María Cecilia Johnson, Fernando Cassorla.
Estradiol and Testosterone effect on the activation of the JAK2/STAT5 pathway induced by growth hormone
1Instituto de Investigación Materno Infantil,
Facultad de Medicina, Universidad de Chile, Santiago-Chile.
Correspondencia:
Paula Ocaranza PhD
pauocaranza@gmail.com
FAX: (56)-2-24247240
Financiamiento: Proyecto SOCHED 2016-03
Recibido: 23-05-2018
Aceptado: 05-06-2018
Abstract: Sex hormones play a major role during pubertal growth. Estradiol (E2) and testosterone (T) levels progressively increase during puberty and in the presence of growth hormone (GH), growth velocity increases. Understanding the interactions between sex hormones and GH, may optimize the treatment of pubertal children with growth disorders. The aim of our study was to investigate possible molecular mechanisms which might potentiate longitudinal growth during puberty due to E2 or T combined with GH. We evaluated the GH/JAK2/STAT5 signaling pathway in the human hepatoma cell line HEPG2. Our results suggest that sex hormones potentiate the GH signaling pathway in a dose dependent fashion. Relatively low concentrations of E2 associated with GH induce a substantial activation of the GH pathway, whereas relatively high concentrations of T associated with GH produce a similar effect. These findings are concordant with the physiology of the pubertal growth spurt, which is an early event in girls (when E2 circulating levels are low), and a late event in boys (when T circulating levels are high).
Key words: Estradiol; Testosterone; Growth hormone; JAK2/STAT5 signaling pathway.
Resumen: Las hormonas sexuales, modulan el crecimiento durante la pubertad. Los niveles de estradiol (E2) y testosterona (T) aumentan progresivamente durante la pubertad y en combinación con la hormona de crecimiento (GH), producen un incremento en la velocidad de crecimiento en este período conocido como el “estirón puberal”. El estudio de la interacción entre las hormonas sexuales y la GH, es de gran importancia para optimizar el tratamiento de niños(as) con alteraciones del crecimiento durante la pubertad. El objetivo de nuestro estudio fue investigar los posibles mecanismos que podrían potenciar el crecimiento longitudinal durante la pubertad, en especial las interacciones entre E2 o T en combinación con GH. Se evaluó la activación de la vía de señalización GH/JAK2/STAT5 frente al estímulo combinado con estas hormonas en cultivos celulares de hepatoma humana HEPG2. Nuestros resultados sugieren que existe un efecto potenciador de las hormonas sexuales sobre la vía de señalización de GH. Observamos que concentraciones relativamente bajas de E2 junto con GH producen una clara activación de la vía de señalización para GH, mientras que concentraciones relativamente altas de T junto con GH producen una activación similar. Estos hallazgos son concordantes con la fisiología del estirón puberal, que es más precoz en niñas (cuando los niveles circulantes de E2 son bajos), y más tardíos en varones (cuando los niveles circulantes de T son altos).
Palabras clave: Estradiol; Testosterona; Hormona de crecimiento; Vía de señalización JAK2/STAT5.
El crecimiento lineal en los seres humanos se caracteriza por una velocidad de crecimiento muy alta durante la etapa fetal, una desaceleración relativa del crecimiento luego del nacimiento, una fase prolongada de crecimiento estable durante la niñez y un estirón puberal durante la adolescencia. La etapa de crecimiento acelerado durante la pubertad depende de la interacción entre el eje somatotrófico y las hormonas sexuales, pero no se han establecido los mecanismos específicos que modulan esta interacción.
La GH es una proteína de 22 kDa, producida por la glándula pituitaria que activa al receptor de hormona de crecimiento (GHR)1. El GHR no presenta actividad quinásica intrínseca, pero el cambio conformacional inducido por la unión del ligando induce la activación de la familia tirosina quinasa asociada a receptores Janus quinasa (JAK)2. La proteína JAK2 se asocia constitutivamente con el GHR, autofosforilándose y fosforilando al receptor. Estas tirosinas fosforiladas forman sitios de unión para varias proteínas como la familia STATs (proteínas transductora de señales y activadoras de la transcripción)3,4. Luego de la unión de GH, las proteínas STATs son reclutadas por el complejo GH/GHR/JAK2, son fosforiladas, produciendo la dimerización de las STATs y su translocación hacia el núcleo, donde se unen al ADN iniciando la transcripción de distintos genes blancos, entre los cuales se encuentra IGF-I4,5,6,7. A pesar que son varios los factores que contribuyen al crecimiento lineal durante la infancia, niñez y la adolescencia, existe consenso que la IGF-I es el principal efector final.
El efecto de las hormonas sexuales como los andrógenos y los estrógenos sobre el crecimiento, es complejo. Los esteroides sexuales pueden tener un efecto directo sobre el crecimiento longitudinal de los huesos y un efecto indirecto sobre la secreción de GH por la glándula pituitaria8. El efecto de los andrógenos sobre la secreción pituitaria de GH es mediada a través de los estrógenos tanto en niñas como en niños8,9. Sin embargo, existe relativamente poca información sobre la posible potenciación de la sensibilidad a GH inducida por los esteroides sexuales durante la pubertad.
El objetivo de este estudio fue investigar el mecanismo molecular mediante el cual se potencia el crecimiento durante la etapa puberal debido a la presencia de las hormonas sexuales en combinación con la hormona de crecimiento. Para responder este objetivo, se evaluó in vitro la activación de JAK2 y STAT5b al estimular con distintas dosis de estradiol o testosterona en presencia de GH en un modelo celular.
Materiales y métodos
Químicos
La hormona de crecimiento recombinante humana (rhGH) fue donada por el Dr. A.F. Parlow (National hormone and peptide program, NIH, USA). El medio Eagle modificado por Dulbecco (DMEM)-baja concentración de glucosa, 17β-estradiol (E2) y testosterona (T) se obtuvieron de Sigma (St. Louis, Mo, USA). El tampón fosfato de Dulbecco (DPBS), penicilina/ estreptomicina y fungizona se adquirieron en Invitrogen (Grand Island, NY, USA). El suero fetal bovino inactivado (SFB) es de BioWest (Biopharm). El estándar de peso molecular se obtuvo de Fermentas (Burlington, Ontario, Canadá).
Cultivos Celulares
Se utilizó la línea celular de hepatoma humana HEPG2 (ATCC CRL-10741) para los distintos ensayos. Los cultivos se propagaron en placas de 60 mm2 en medio DMEM suplementado con SFB al 10%, penicilina 50 IU/mL, estreptomicina 50 mg/ mL y fungizona 0,25 mg/mL. Los cultivos se mantuvieron a 37°C en una atmósfera de 5% CO2 hasta su uso.
Estudios de tiempo-respuesta de las hormonas esteroidales y de hormona de crecimiento
Se realizaron estudios de tiempo-respuesta para cada hormona que consistió en una preincubación inicial con la hormona esteroidal (E2 o T) durante 24h seguido de una coestimulación durante 15 min en presencia de la hormona esteroidal más rhGH (40 ng/mL). Estudios realizados en nuestro laboratorio en cultivos de fibroblastos de piel muestran que son necesarias concentraciones de rhGH de al menos 200 ng/mL para activar la vía de señalización de GH en este modelo celular, modelo poco sensible a la estimulación con fhGH10. La células HEPG2 es una buena respondedora al tratamiento con rhGH, se necesitan menores concentraciones para estimular la vía GH/JAK2/STAT5b10,11. Las concentraciones de E2 estudiadas fueron 0, 20, 50, 100 y 200 pg/mL, estas concentraciones bajas son las observadas en niñas prepuberales y las altas en mujeres post menarquia. Las concentraciones de T estudiadas fueron 0, 1, 2.5, 5 y 10 ng/mL y estas concentraciones se establecieron considerando niveles observados en niños durante la transición de prepubertad a adolescencia.
Extracción de proteínas citoplasmáticas
Los cultivos de HEPG2 se privaron de suero y se estimularon con distintas concentraciones de hormonas esteroidales durante 24h en presencia o ausencia de la coestimulación con rhGH. Los extractos de la fracción citoplasmática se prepararon según lo descrito por Schreiber y cols. (1989)12 y el contenido proteico se determinó con el ensayo de Bradford (1976)13.
Análisis del contenido proteico mediante inmunoblots
Los extractos proteicos se resolvieron mediante SDSPAGE al 10% bajo condiciones reductoras y se transfirieron a membranas de nitrocelulosa (Bio-Rad, Richmond, USA). Las membranas se bloquearon con BSA 3% durante 1h para luego incubar a 4°C por 16h con el anticuerpo correspondiente. Luego de varios lavados, se incubó 1h con el anticuerpo secundario correspondiente y las proteínas se visualizaron por quimioluminiscencia (Pierce ECL Western Blotting Substrate Kit; Pierce, Rockford, IL, USA) utilizando el equipo Discovery 10gD Ultralum con el software Ultraquant (versión 6.0.0.344). Las intensidades de las bandas se cuantificaron usando el programa Image J 1.38x (NIH). Los anticuerpos utilizados fueron: anti-fosfo-STAT5-Tyr694 1:2500, anti-fosfo-JAK2-Tyr1007/1008 1:2000 and anti-JAK2 1:2000 (Cell Signaling Technology de Danvers, MA, USA); anti-STAT5 1:2000 (Boster Biological Technology de Pleasanton, CA, USA), y anti-β-actina 1:100.000 (Sigma Aldrich) utilizado como control interno. Los anticuerpos secundarios anti-rabbit IgG o anti-mouse IgG conjugados con peroxidasa fueron de Rockland 1:10.000 (Gilbertsville, PA, USA).
Análisis estadístico
Los resultados se expresaron como el promedio±EEM. Los datos se compararon con respecto a su basal mediante los análisis estadísticos de Kruskal Wallis seguido por Dunn. El n para cada estímulo fue de al menos 5. Para el análisis de los datos se utilizó el Programa SPSS v21 y los valores de P <0,05 se consideraron como significativos.
Resultados
Activación de la vía de señalización de hormona de crecimiento en células HEPG2
Para evaluar la activación de la vía de señalización de GH, cultivos de HEPG2 se estimularon con una concentración única de rhGH correspondiente a 40 ng/mL durante 15 min. Transcurrido el tiempo de estimulación, se evaluó mediante inmunoblot los niveles de fosforilación de JAK2 y STAT5 en el citoplasma. En la Figura 1A se observa un aumento en los niveles de fosforilación de JAK2 en comparación a la condición sin estímulo (0,34±0,1 unidades arbitrarias (UA) vs 0,77±0,06 UA; *p<0,05). Resultados similares se obtuvieron para STAT5 frente al estímulo con rhGH (Figura 1B) (0,34±0,1 UA vs 0,90±0,06 UA; *p< 0,05).
Efecto de estradiol y testosterona sobre la fosforilación de JAK2 y STAT5 en células HEPG2
Se estimularon las células con concentraciones crecientes de E2 entre 20-200 pg/mL durante 24h y los resultados obtenidos se compararon con la condición basal (ausencia de E2). Se puede observar que tanto JAK2 (Figura 2A) como STAT5 (Figura 2B) no modificaron sus niveles de fosforilación al estimular con E2. Al evaluar el posible efecto de T en concentraciones crecientes entre 1-10 ng/mL sobre la activación de JAK2 (Figura 3A) y de STAT5 (Figura 3B) se observó que tampoco se modificaron al estimular sólo con T. Estos resultados indican que tanto el E2 como la T en forma aislada no producen una activación de la vía de señalización JAK2/STAT5 asociada a GH en células HEPG2.
Efecto combinado de hormona de crecimiento más estradiol o testosterona sobre la activación de JAK2 y STAT5b en células HEPG2
Teniendo como antecedente que el E2 y la T solas no son capaces de activar directamente la vía JAK/STAT, cultivos de células HEPG2 se preincubaron con distintas concentraciones de E2 o T durante 24h para luego agregar una concentración de 40 ng/mL de rhGH durante 15 minutos.
La figura 4 muestra el efecto de la combinación de estímulos de E2 más rhGH (E2+rhGH) sobre la activación de JAK2 y STAT5b. Al estimular con E2+rhGH se evidenció un aumento en los niveles de fosforilación de JAK2 (Figura 4A) con todas las concentraciones de E2 estudiadas en comparación al estímulo único de rhGH (0,82±0,006 UA). Es importante destacar que el mayor nivel de fosforilación de JAK2 se observó la concentración E2+GH menor de E2 utilizada (20 pg/mL; 1,52±0,14 UA vs 0,82±0,006 UA; *p< 0,05). La figura 4B muestra un aumento en los niveles de fosforilación de STAT5 citoplasmática al estimular con rhGH; sin embargo, este efecto no se incrementó al preincubar con distintas concentraciones de E2.
Por otro lado, al preincubar los cultivos con concentraciones crecientes de T más rhGH, se observó que los niveles de fosforilación de JAK2 se incrementaron a medida que aumentan las concentraciones de T (Figura 5A), alcanzando diferencia significativa con las concentraciones de T de 5 ng/mL (1,35±0,23 UA vs 0,87±0,03 UA) y 10 ng/mL (1,85±0,21 UA vs 0,87±0,03 UA). Además, la fosforilación de STAT5 citoplasmática (Figura 5B) aumentó sólo ante la presencia de rhGH y la más alta concentración de T (10 ng/mL) en comparación a rhGH sola (0,82±0,04 UA vs 1,32±0,02 UA *p<0,05).
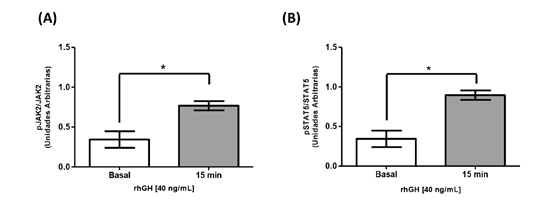 Figura 1. Activación de la vía de hormona de crecimiento en células HEPG2 mediante la estimulación con rhGH. Cultivos de células HEPG2 se estimularon durante 15 minutos con rhGH [40 ng/mL]. Las proteínas se estudiaron utilizando la técnica de inmunoblot (A) Cuantificación de la fosforilación de JAK2 en células HEPG2 en respuesta a rhGH. (B) Cuantificación de la fosforilación de STAT5 en células HEPG2 en respuesta a rhGH. Los valores se expresan como prom ± EEM. *p< 0,05 (n= 5).
Figura 1. Activación de la vía de hormona de crecimiento en células HEPG2 mediante la estimulación con rhGH. Cultivos de células HEPG2 se estimularon durante 15 minutos con rhGH [40 ng/mL]. Las proteínas se estudiaron utilizando la técnica de inmunoblot (A) Cuantificación de la fosforilación de JAK2 en células HEPG2 en respuesta a rhGH. (B) Cuantificación de la fosforilación de STAT5 en células HEPG2 en respuesta a rhGH. Los valores se expresan como prom ± EEM. *p< 0,05 (n= 5). 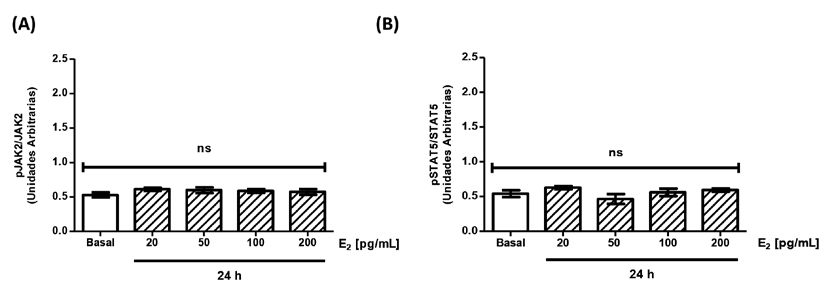 Figura 2. Activación de la vía de hormona de crecimiento en células HEPG2 mediante la activación de estradiol. Cultivos de células HEPG2 se estimularon
durante 24 h con concentraciones crecientes de E2. Transcurrido el tiempo de estimulación, las proteínas se estudiaron utilizando la técnica de inmunoblot. (A)
Cuantificación de la fosforilación de JAK2 en células HEPG2 en respuesta a E2. (B) Cuantificación Cuantificación de la fosforilación de STAT5 en células HEPG2 en respuesta a
E2. Los valores se expresan como prom ± EEM. ns= no significativo (n= 5).
Figura 2. Activación de la vía de hormona de crecimiento en células HEPG2 mediante la activación de estradiol. Cultivos de células HEPG2 se estimularon
durante 24 h con concentraciones crecientes de E2. Transcurrido el tiempo de estimulación, las proteínas se estudiaron utilizando la técnica de inmunoblot. (A)
Cuantificación de la fosforilación de JAK2 en células HEPG2 en respuesta a E2. (B) Cuantificación Cuantificación de la fosforilación de STAT5 en células HEPG2 en respuesta a
E2. Los valores se expresan como prom ± EEM. ns= no significativo (n= 5).
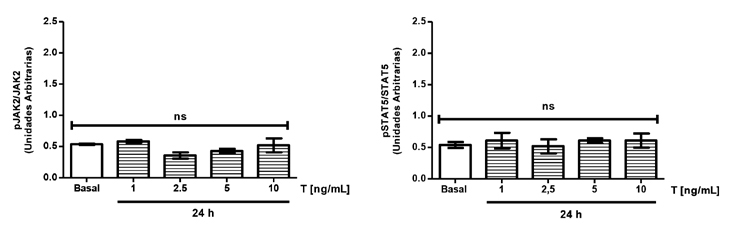 Figura 3. Activación de la vía de hormona
de crecimiento en células HEPG2 mediante
la activación de testosterona. Cultivos de
células HEPG2 se estimularon durante 24 h con
concentraciones crecientes de T. Transcurrido
el tiempo de estimulación, las proteínas se
estudiaron utilizando la técnica de inmunoblot.
(A) Cuantificación de la fosforilación de JAK2
en células HEPG2 en respuesta a T. (B)
Cuantificación de la fosforilación de STAT5
en células HEPG2 en respuesta a T. Los
valores se expresan como prom ± EEM. ns =
no significativo (n= 5).
Figura 3. Activación de la vía de hormona
de crecimiento en células HEPG2 mediante
la activación de testosterona. Cultivos de
células HEPG2 se estimularon durante 24 h con
concentraciones crecientes de T. Transcurrido
el tiempo de estimulación, las proteínas se
estudiaron utilizando la técnica de inmunoblot.
(A) Cuantificación de la fosforilación de JAK2
en células HEPG2 en respuesta a T. (B)
Cuantificación de la fosforilación de STAT5
en células HEPG2 en respuesta a T. Los
valores se expresan como prom ± EEM. ns =
no significativo (n= 5).
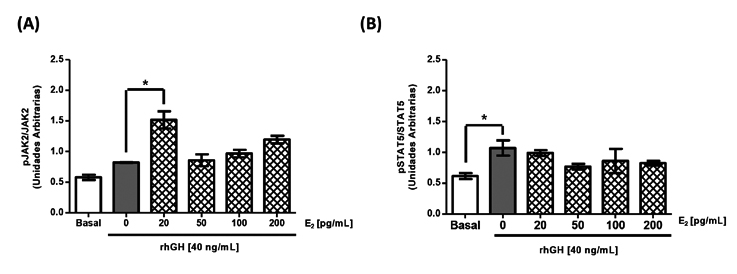 Figura 4. Efecto de la preincubación de
estradiol y hormona de crecimiento sobre
la vía transducciónal de GH. Cultivos de
células HEPG2 se estimularon durante 24 h
con concentraciones crecientes de E2 y luego
coincubaron durante 15 min con E2 más rhGH.
Transcurrido el tiempo de estimulación, las
proteínas se estudiaron utilizando la técnica de
inmunoblot. (A) Cuantificación de la fosforilación
de JAK2 en células HEPG2 en respuesta a la
coincubación de E2 y rhGH. (B) Cuantificación
de la fosforilación de STAT5 en células HEPG2
en respuesta a la coincubación de E2 y rhGH.
Los valores se expre
Figura 4. Efecto de la preincubación de
estradiol y hormona de crecimiento sobre
la vía transducciónal de GH. Cultivos de
células HEPG2 se estimularon durante 24 h
con concentraciones crecientes de E2 y luego
coincubaron durante 15 min con E2 más rhGH.
Transcurrido el tiempo de estimulación, las
proteínas se estudiaron utilizando la técnica de
inmunoblot. (A) Cuantificación de la fosforilación
de JAK2 en células HEPG2 en respuesta a la
coincubación de E2 y rhGH. (B) Cuantificación
de la fosforilación de STAT5 en células HEPG2
en respuesta a la coincubación de E2 y rhGH.
Los valores se expre
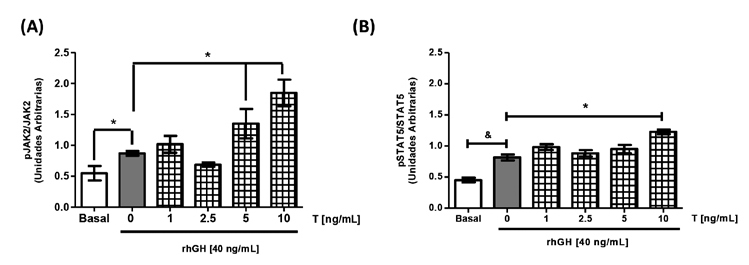 Figura 5. Efecto de la preincubación de
testosterona y hormona de crecimiento sobre
la vía transducciónal de GH. Cultivos de
células HEPG2 se estimularon durante 24 h
con concentraciones crecientes de T y luego
coincubaron durante 15 min con T más rhGH.
Transcurrido el tiempo de estimulación, las
proteínas se estudiaron utilizando la técnica de
inmunoblot. (A) Cuantificación de la fosforilación
de JAK2 en células HEPG2 en respuesta a la
coincubación de T y rhGH. (B) Cuantificación
de la fosforilación de STAT5 en células HEPG2
en respuesta a la coincubación de T y rhGH.
Los valores se expresan como prom ± EEM.
*p< 0,05 (n= 5)..
Figura 5. Efecto de la preincubación de
testosterona y hormona de crecimiento sobre
la vía transducciónal de GH. Cultivos de
células HEPG2 se estimularon durante 24 h
con concentraciones crecientes de T y luego
coincubaron durante 15 min con T más rhGH.
Transcurrido el tiempo de estimulación, las
proteínas se estudiaron utilizando la técnica de
inmunoblot. (A) Cuantificación de la fosforilación
de JAK2 en células HEPG2 en respuesta a la
coincubación de T y rhGH. (B) Cuantificación
de la fosforilación de STAT5 en células HEPG2
en respuesta a la coincubación de T y rhGH.
Los valores se expresan como prom ± EEM.
*p< 0,05 (n= 5).. Discusión
El crecimiento postnatal en humanos presenta etapas independientes (prenatal) y dependientes (postnatal) de GH14. La GH secretada desde la pituitaria de manera género dependiente se traduce en un patrón de secreción que lleva a diferencias en la expresión de genes principalmente de STAT5b15. Además, la producción de GH y su liberación se encuentra significativamente afectada por las hormonas sexuales. De hecho, los niveles de GH y estrógenos muestran una asociación positiva tanto en niñas como niños prepuberales16.
En este estudio, exploramos el posible efecto potenciador de E2 o T sobre la capacidad de GH para activar la vía JAK2/ STAT5 en una línea celular hepática humana. Esta línea celular presenta todos los componentes de la vía de señalización de GH, por lo que JAK2/STAT5b son rápidamente activados cuando son estimulados con rhGH.
En el presente trabajo encontramos que al estimular estas células durante 24 horas con concentraciones relativamente bajas de E2 más 15 min con rhGH, la activación de JAK2 aumenta un 85% en comparación a la inducida por rhGH sola. Por otro lado, la activación de JAK2 aumenta un 112% al estimular con rhGH y concentraciones altas de T en comparación con el estímulo con rhGH sola. Nuestros resultados sugieren que existiría un efecto potenciador de las hormonas sexuales sobre la vía de señalización de GH ya que la estimulación única con E2 o T, incluso a altas concentraciones de cada una de estas hormonas no modifican la fosforilación de JAK2 o de STAT5b.
La estimulación con E2 o T seguido de una coincubación con rhGH activó a STAT5b citoplasmático a niveles de fosforilación similares a los observados con la estimulación de rhGH por sí sola. Esta mantención en la forma fosforilada de STAT5b, podría indicar que luego de la activación de STAT5b en el citoplasma, éste es rápidamente translocado hacia el núcleo actuando como factor de transcripción para genes específicos como IGF-I e IGFBP-3s17,18. Estos resultados plantean la necesidad de estudiar la activación de STAT5b nuclear inducida por la preincubación con E2 a bajas concentraciones y el posterior estímulo con rhGH.
Al finalizar la pubertad, las altas concentraciones sistémicas de GH encontradas durante esta etapa disminuyen a concentraciones similares a las observadas durante la prepubertad independientemente de las concentraciones crecientes de las hormonas sexuales19. Estos antecedentes indican que existen concentraciones críticas de E2 presentes durante la pubertad, que aumentarían la sensibilidad tisular a GH. Por otro lado, en niños con pubertad precoz tratados con análogos de GnRH, se produce una inhibición de la secreción de esteroides sexuales por las gónadas y de GH por la glándula pituitaria, lo que reduce los niveles circulantes de IGF-I20,21. Nuestros resultados sugieren que la reducción de los niveles de esteroides sexuales podría además afectar la sensibilidad tisular a GH, lo que podría afectar el crecimiento lineal de los pacientes con pubertad precoz tratados con análogos de GnRH.
Existe evidencia que concentraciones altas de E2pueden acelerar el cierre de la placa de crecimiento sin incrementar la velocidad de crecimiento22 mientras que concentraciones bajas de E2 favorecen el crecimiento lineal23. Esta literatura concuerda con nuestros resultados obtenidos con una concentración relativamente baja de E2 (20 pg/mL, observado en niñas durante el inicio de la pubertad), induce una mayor activación de la vía JAK2/STAT5. Por otro lado, una concentración relativamente alta de E2 (200 pg/mL, observado en mujeres adultas), favorece la activación de la vía JAK2/STAT5 inducida por GH. Nuestros datos indican que se requieren concentraciones relativamente bajas de E2 junto con GH para una mayor activación de la vía de señalización de GH en nuestra línea celular.
Por otro lado, el efecto de T sobre esta vía de señalización intracelular se observa preferentemente con concentraciones relativamente elevadas de este esteroide. Cabe mencionar que estas concentraciones de T pueden inducir este efecto sobre la vía JAK2/STAT5 debido a su conversión a E2 por acción de la enzima aromatasa. En este sentido, sería útil estudiar el efecto de un andrógeno no aromatizable como la dihidrotestosterona sobre esta vía de señalización intracelular para GH.
Existe escasa información sobre la posible modulación de la sensibilidad tisular para GH inducida por los esteroides sexuales. Uno de ellos, utilizó la línea celular renal humana HEK293 que no expresa constitutivamente los receptores para GH y E224, por lo que debieron transfectar las células. Los autores demostraron que el E2 en concentraciones suprafisiológicas (272-27200 pg/mL) induce la expresión de SOCS2 lo que inhibe la señalización de JAK2/STAT5b24. Un estudio posterior realizado en la línea celular de hepatoma humana HuH7, utilizando las mismas concentraciones suprafisiológicas de E2, indicó que tanto SOCS2 como SOCS3 son inducidos frente al estímulo con E2 más GH, inhibiendo la vía de GH25. Por el contrario, nosotros encontramos un efecto potenciador a concentraciones relativamente bajas de E2 sobre la vía de señalización de JAK/STAT5b inducida por GH.
Sugerimos como mediador clave entre las vías de señalización GH/E2(T) a la proteína SOCS2, proteína que tendría una función dual según la concentración de E2 presente en el medio. A bajas concentraciones de E2 coincubadas con rhGH, SOCS2 podría estar favoreciendo la vía de señalización de JAK2/ STAT5 debido a la inhibición directa sobre SOCS3 descrito en líneas celulares HEK293-T y 3T3-F442A26,27. Por otro lado, frente a concentraciones altas de E2 coincubadas con rhGH, el aumento en el contenido proteico de SOCS2 inhibiría la vía de GH evitando la fosforilación de JAK2 y permitiría la acción de SOCS3 que se ha descrito como un inhibidor más potente de la vía de señalización de GH28,29.
En resumen, nuestros resultados indican que concentraciones relativamente bajas de E2 y relativamente altas de T modularían la sensibilidad tisular a GH, particularmente en la vía de señalización GH/JAK2/STAT5 en células HEPG2. Estos hallazgos son concordantes con la fisiología del estirón puberal, que es más precoz en niñas (cuando los niveles circulantes de E2 son bajos), y más tardíos en varones (cuando los niveles circulantes de T son más altos).
Referencias
- Radetti G, Bozzola M, Pagani S, Avanzini MA, Messner H, Belloni C, Hayakawa M. Circulating GH isoforms and GH bioactivity in preterm neonates. Pediatr Res 2000; 48: 244-247.
- Zhang Y, Jiang J, Kopchick JJ, Frank SJ. “Disulfide linkage of growth hormone (GH) receptors (GHR) reflects GH-induced GHR dimerisation. Association of JAK2 with the GHR is enhanced by receptor dimerisation.” J Biol Chem 1999; 274: 33072-33084.
- Woelfle J, Billiard P, Rotwein P. Acute control of insulin-like growth factor-I gene transcription by growth hormone through STAT5b. J Biol Chem 2003; 278: 22696-22702.
- Davey HW, Xie T, McLachlan MJ, Wilkins RJ, Waxman DJ, Grattan DR. STAT5b is required for GH-induced liver IGF-I gene expression. Endocrinol 2001; 142: 3836-3841.
- Carter-Su C, Schwartz J, Smit LS. Molecular mechanism of growth hormone action. Annu Rev Physiol 1996; 58: 187-207.
- Woelfle J, Chia DJ, Rotwein P. Mechanisms of growth hormone (GH) action. Identification of conserved Stat5 binding sites that mediate GHinduced insulin-like growth factor-I gene activation. J Biol Chem 2003; 278: 51261-51266.
- Davey HW, Xie T, McLachlan MJ, Wilkins RJ, Waxman DJ, Grattan DR. STAT5b is required for GH-induced liver IGF-1 gene expression. Endocrinol 2001; 142: 3836-3841.
- Perry RJ, Farquharson C, Ahmed SF. The role of sex steroids in controlling pubertal growth. Clin Endocrinol 2008; 68: 4-15.
- Veldhuis JD, Metzger DL, Martha PM Jr., Mauras N, Kerrigan JR, Keenan B, Rogol AD, Pincus SM. Estrogen and Testosterone, but not a nonaromatizable androgen, direct network integration of the hypothalamo-somatotrope (growth hormone)-insulin-like growth factor I axis in the human: Evidence from pubertal pathophysiology and sex-steroid hormone replacement. JCEM 1997; 82: 3414-3420.
- Ocaranza P, Morales F, Matamala Á, Gaete X, Román R, Lammoglia JJ, et al. Growth hormone signaling in fibroblasts from newborn boys and prepubertal boys. Growth Horm IGF Res 2016; 27: 18-27.
- Silva CM, Kloth MT, Whatmore AJ, Freeth JS, Anderson N, Laughlin KK, et al. GH and epidermal growth factor signaling in normal and Laron syndrome fibroblasts. Endocrinol 2002; 143: 2610-2617.
- Schreiber E, Matthias P, Müller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini extracts’, prepared from a small number of cells. Nucleic Acids Res 1989; 17: 6419.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976; 72: 248-254.
- Pagani S, Chaler EA, Radetti G, Travaglino P, Meazza C, Bozzola E, et al. Variations in biological and immunological activity of growth hormone during the neonatal period. Horm Res 2007; 68: 145-149.
- Clodfelter KH, Holloway MG, Hodor P, Park S-H, Ray WL, Waxman DJ. Sex-dependent liver gene expression is extensive and largely dependent upon signal transducer and activator of transcription 5b (STAT5b): STAT5bdependent activation of male genes and repression of female genes revealed by microarray analysis. Mol Endocrinol 2006; 20: 1333-1351.
- Veldhuis JD, Roemmich JN, Rogol AD. Gender and sexual, maturationdependent contrasts in the neuroregulation of growth hormone secretion in prepubertal and late adolescent males and females-A general clinical research center-based study. J Clin Endocrinol Metab 2000; 85: 2385- 2394.
- Woelfle J, Rotwein P. In vivo regulation of growth hormone-stimulated gene transcription by STAT5b. Am J Physiol Endocrinol Metab 2004; 286: E393-E401
- Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med 2003; 349: 1139-1147.
- Martha PM Jr, Rogol AD, Veldhuis JD, Kerrigan JR, Goodman DW, Blizzard RM. Alterations in the pulsatile properties of circulating growth hormone concentrations during puberty in boys. J Clin Endocrinol Metab 1989; 69: 563-570.
- Mericq MV, Eggers M, Avila A, Cutler GB Jr., Cassorla F. Near final height in pubertal growth hormone (GH)-deficient patients treated with GH alone or in combination with luteinizing hormone-releasing hormone analog: results of a prospective, randomized trial. J Clin Endocrinol Metab 2000; 85: 569-573.
- Attie KM, Ramirez NR, Conte FA, Kaplan SL, Grumbach MM. The pubertal growth spurt in eight patients with true precocious puberty and growth hormone deficiency: evidence for a direct role of sex steroids. J Clin Endocrinol Metab 1990; 71: 975-983.
- Drop SL, de Waal WJ, de Muinck Keizer-Schrama SM. Sex steroid treatment of constitutionally tall stature. Endocrine Rev 1998; 19: 540-558.
- Ross JL, Cassorla FG, Skerda MC, Valk IM, Loriaux DL, Cutler GB. A preliminary study of the effect of estrogen dose on growth in Turner’s Syndrome. N Engl J Med 1983; 309: 1104-1106.
- Leung KC, Doyle N, Ballesteros M, Sjorgen K, Watts CKW, Low TH, et al. Estrogen inhibits GH signaling by suppressing GH-induced JAK2 phosphorylation, an effect mediated by SOCS-2. Proc Nat Acad Sci 2003; 100: 1016-1021.
- Leong GM, Moverare S, Brce J, Doyle N, Sjögren K, Dahlman-Wright K, et al. Estrogen up-regulates hepatic expression of suppressors of cytokine signaling-2 and -3 in vivo and in vitro. Endocrinol 2004; 145: 5525-5531.
- Piessevaux J, Lavens D, Montoye T, Wauman J, Catteeuw D, Vandekerckhove J, et al. Functional cross-modulation between SOCS proteins can stimulate cytokine signaling. J Biol Chem 2006; 281: 32953-32966.
- Piessevaux J, Lavens D, Peelman F, Tavernier J. The many faces of the SOCS box. Cytokine Growth Factor Rev 2008; 19: 371-381.
- Schaefer F, Chen Y, Tsao T, Nouri P, Rabkin R. Impaired JAK-STAT signal transduction contributes to growth hormone resistance in chronic uremia. J Clin Invest 2001; 108: 467-475.
- Ocaranza P, Morales F, Román R, Iñiguez G, Fernando C. Expression of SOCS1, SOCS2, and SOCS3 in growth hormone-stimulated skin fibroblasts from children with idiopathic short stature. J Pediatr Endocrinol Metab 2012; 25: 273-278.