Acromegalia y silla turca vacía. Una asociación infrecuente. Presentación de un caso
Irasel Martínez Montenegro1*, Claudia Borges Alonso2.
Acromegaly and empty sella syndrome. An infrequent association. Presentation of a case
1 Especialista en Endocrinología. Máster en Infectología. Profesor Instructor. Servicio Endocrinología Hospital Teodoro Maldonado Carbo. Guayaquil, Guayas, Ecuador.
2 Especialista en Endocrinología. Servicio Endocrinología Hospital General del Norte de Guayaquil Los Ceibos. Guayaquil, Guayas, Ecuador.
* Correspondencia: Irasel Martínez Montenegro / irasel051283@gmail.com. Teléfono 0989464147.
Recibido: 13-01-2019.
Aceptado: 25-04-2019.
Resumen: La acromegalia, originada por un exceso de producción de Hormona de crecimiento (Gh), se caracteriza por crecimiento somático exagerado, alto riesgo cardio-metabólico, así como reducción de la expectativa de vida. Tiene una incidencia de 3-4 casos por millón de habitantes. El diagnóstico se retrasa hasta 10 años aumentando la morbi-mortalidad.
Las alternativas terapéuticas incluyen medicamentos y cirugía, que van encaminados a reducir los efectos de masa tumoral, normalizar los parámetros bioquímicos y resolver las manifestaciones clínicas. En casos muy infrecuentes, el tumor hipofisario que la origina se asocia a silla turca vacía.
Palabras clave: Acromegalia, Silla turca vacía, Tumor hipofisario.
Abstract: Acromegaly, caused by an excess production of growth hormone (Gh), it is characterized by exaggerated somatic growth, high cardio-metabolic risk, as well as reduction of life expectancy. It has an incidence of 3-4 cases per million population. The diagnosis
is delayed up to 10 years increasing morbidity and mortality. The therapeutic alternatives include medications and surgery, which are aimed at reduce the effects of tumor mass, normalize biochemical parameters and resolve clinical manifestations. In very infrequent cases, the pituitary tumor that originates it is associated with empty sella syndrome.
Key words: Acromegaly, Empty sella syndrome, Pituitary tumor.
La gran mayoría de los pacientes con diagnóstico de Acromegalia presentan un adenoma productor de GH (Hormona de crecimiento) como causa. Por lo tanto, es importante identificar y tratar adecuadamente a los pacientes con características de acromegalia y niveles elevados de IGF-1 (insulin-like growth factor-1), incluso si sus concentraciones de GH en plasma son aparentemente normales1.
En ocasiones puede acompañarse de silla turca vacía, consecuencia de un infarto en un adenoma hipofisario previo, estos pacientes suelen tener tejido hipofisario comprimido contra el suelo de la silla turca. Aproximadamente un 10% de los pacientes presentarán adenomas pequeños secretores de GH o PRL (Prolactina), dentro del estrecho ribete de tejido hipofisario comprimido2.
Caso clínico
Paciente femenina de 69 años de edad con: antecedente quirúrgico de colecistectomía; antecedentes patológicos personales de obesidad, hipertensión arterial esencial, osteoporosis, diverticulosis colónica y amiloidosis. Además, diabetes mellitus tipo 2 (DM2) con complicaciones microangiopáticas, tratada con insulina de acción intermedia y metformina.
Acude a consulta de endocrinología refiriendo dolores articulares generalizados y estudio anterior, por sospecha de acromegalia, iniciado en 2015. Al examen físico se aprecia: fascie acromegaloidea con nariz y labios grandes y carnosos.
En exámenes de laboratorio se obtuvieron los siguientes resultados: IGF-1 372 ng/ml (69 - 200 ng/mL); prueba de tolerancia oral a la glucosa midiendo GH (PTG-GH): 0 minutos - 2,14/ 30 minutos - 1,38/ 60 minutos - 1,49/ 90 minutos - 1,52/ 120 minutos - 1,52/ 150 minutos - 1,55/ 180 minutos - 1,21 ng/ml; TSH: 1.28 ulU/ml (0.4 - 4 ulU/ml); prolactina: 5.2 ng/ mL (1.9 - 25 ng/mL). Los estudios de imagen informaron: RMN (Resonancia magnética nuclear) de hipófisis región selar parcialmente llena de líquido cefalorraquídeo (LCR) con dificultad para identificar el tejido hipofisario, herniación de aracnoides y el LCR que comprime a la glándula contra el suelo selar; tras la administración de contraste solo se aprecia refuerzo del infundíbulo, tallo hipofisario en línea media de aspecto normal, quiasma óptico de características normales, estructura hipotalámica normal, asimetría en la relación de los diámetros mayores entre el macizo cráneo facial y la bóveda craneal (Figura 1a y 1b). En Radiografía (Rx) columna: Rotoescoliosis, acentuación de la lordosis lumbar, disminución de espacios discales lumbares. Rx rodilla: prominencia de las espinas tibiales medial y lateral, disminución del espacio interarticular femoropatelar y femorotibial interno, osteofitos en cóndilo femoral y tibial. Rx de mano: erosiones óseas marginales, estrechamiento de los espacios interfalangicos.
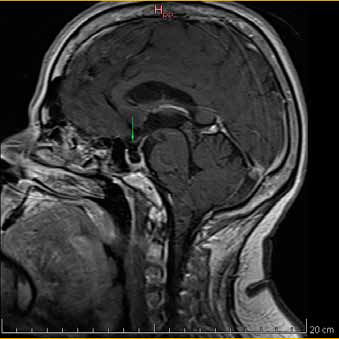 Figura 1a. RMN de silla turca contrastada con gadolinio. Corte sagital en T1. Cortes 2.5 mm. Se señala región selar.
Figura 1a. RMN de silla turca contrastada con gadolinio. Corte sagital en T1. Cortes 2.5 mm. Se señala región selar.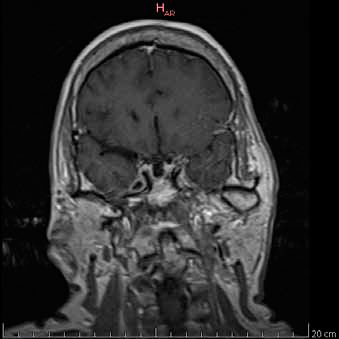 Figura 1b. RMN de silla turca contrastada con gadolinio. Corte coronal en T1. Cortes 2.5 mm.
Figura 1b. RMN de silla turca contrastada con gadolinio. Corte coronal en T1. Cortes 2.5 mm.Rx de tórax: diámetro transverso del corazón aumentado. Teniendo en cuenta el cuadro clínico, los estudios hormonales y radiológicos, se realizó el diagnóstico de acromegalia. Se indicó el tratamiento farmacológico con octreotide 20 mg 1 bulbo mensual, logrando mejoría de la sintomatología dolorosa, control de DM2 e hipertensión arterial y, en los exámenes de laboratorio un valor de IGF-1 igual a 173 ng/ml (69 - 200 ng/mL).
Discusión
El diagnóstico de acromegalia requiere la realización de una Prueba de sobrecarga oral con 75 g de glucosa midiendo a la vez GH; valores superiores a 0,4 ng/ml en dicho valor, utilizando análisis ultrasensibles, o más de 1 ng/ml empleando métodos estándar, confirman el diagnóstico, con una concentración de IGF-1 concomitante elevada3,4.
El contenido basal de GH determina los valores de IGF-1 circulantes, según un patrón logarítmico- lineal5. La detección de características clínicas singulares o imprevistas como: estertores respiratorios o disnea, rubor facial, úlceras pépticas o cálculos renales, hipoglucemia, hiperinsulinemia, la hipergastrinemia y, raras veces, el hipercortisolismo, justifican la evaluación de una posible fuente extrahipofisaria de exceso de GH. La RMN y la TAC (tomografía axial computarizada) pueden ser utilizadas como medio diagnóstico en la localización de tumores tanto hipofisarios como extrahipofisarios2.
Cuando las características clínicas de la acromegalia se asocian a GH e IGF-1 normales, cabe considerar una posible acromegalia “agotada” o “asintomática”, asociada a un adenoma hipofisario infartado, a menudo en espacio de silla turca vacía1.
En la paciente evaluada no puede considerarse el diagnóstico de acromegalia “agotada”, ya que presenta actividad bioquímica. Incluso la actividad bioquímica mínima de la acromegalia puede asociarse con un síndrome clínico grave y completo, como se refleja en los resultados de PTG midiendo GH, que alcanzan valores no muy superiores a los de referencia, en cambio el cuadro clínico de la paciente muestra los signos completos descritos en la enfermedad.
Las masas hipofisarias pueden sufrir un infarto asintomático, con aparición de espacio vacío parcial o total en la silla turca, con una reserva hipofisaria normal, lo que implica que el ribete circundante de tejido hipofisario tiene funcionabilidad normal. En ocasiones poco frecuentes aparecen adenomas hipofisarios funcionales sobre el resto de tejido hipofisario, no visibles en la RMN (menos de 2 mm de diámetro), a pesar de su hiperactividad endocrina. Por lo tanto, estamos ante la presencia de una paciente con diagnóstico de Acromegalia con causa en un tumor hipofisario productor de GH, que se presenta en el contexto de una silla turca vacía, probablemente primaria.
Referencias
- Dimaraki EV, Jaffe CA, DeMott-Friberg R, et al. Acromegaly with apparently norm al GH secretion; implications for diagnosis and follow-up. J Clin Endocrinol Metab 2002; 87: 3537-3542
- Melmed S, Kleinberg D. Masas y tumores hipofisario. En: Williams, 13 ed. Tratado de Endocrinología. Editorial Elsevier 2017; 232- 276.
- Freda PU, Reyes CM, Nuruzzaman AT, et al. Basal and glucose-suppressed GH levels less than 1 microg/L in newly diagnosed acromegaly. Pituitary. 2003; 6: 175-180.
- Ribeiro-Oliveira Jr A, Barkan A. The changing face of acromegaly-advances in diagnosis and treatment. Nat Rev Endocrinol. 2012; 8: 605-611.
- Faje AT, Barkan AL. Basal, but not pulsatile, growth hormone secretion determines the ambient circulating levels of insulin-like growth factor-I. J Clin Endocrinol Metab. 2010; 95: 2486-2491.