Síndrome de Ovario Poliquístico: nuevas perspectivas
Teresa Sir Petermann.
New perspectives on polycystic ovary syndrome.
Profesora Titular de Medicina, Universidad de Chile. Laboratorio de Endocrinología y Metabolismo, Unidad de Endocrinología, Facultad de Medicina Occidente, Universidad de Chile, Hospital San Juan de Dios.
Correspondencia a: Teresa Sir Petermann Laboratorio de Endocrinología y Metabolismo Facultad de Medicina Occidente, Universidad de Chile. Las Palmeras 299, Quinta Normal (Interior) e-mail: tsir@med.uchile.cl.
Recibido el 01 de septiembre, 2008. Aceptado el 08 de septiembre, 2008.
Currently there are two definitions for polycystic ovary syndrome (PCOS). One was proposed by the NIH Consensus Conference (1990), which defined it as the presence of hyperandrogenism and oligo-ovulation in the absence of other specific causes of ovarian, adrenal, or hypophyseal origin. The other proposed by the Rotterdam ESHRE/ASRM Consensus Conference (2003), which incorporates ovarian morphology to the definition generating four different phenotypes. Most women with PCOS exhibit peripheral insulin resistance, which plays a major role in the long-term metabolic consequences of the syndrome, among which type 2 diabetes and cardiovascular disease should be highlighted. Therefore, this syndrome is currently defined as a mainly endocrine metabolic disorder. PCOS could have a genetic predisposition that may manifest itself even before menarche. On the other hand, environmental factors during prenatal or postnatal life could lead to clinical and biochemical expressions of the syndrome in adult life. At present, it is accepted that prenatal exposure to androgens during fetal life is implicated in the origin of this syndrome. We have demonstrated that PCOS mothers may offer a hyperandrogenic and hyperinsulinemic milieu to the female and male fetus. This adverse intrauterine milieu could induce fetal programming consequences, which are evidenced by a high frequency of low-birth weight infants and changes in the testicular and ovarian function. Moreover, some of the metabolic features of PCOS are present in the offspring of PCOS women. Therefore, these children are a high risk group for metabolic and reproductive alterations. Consequently the treatment for this syndrome must not only be symptomatic but also fundamentally preventive. For this reason, PCOS should be considered a marker of a family pathology, a pathway to type 2 diabetes, and a public health problem.
El Síndrome de Ovario Poliquístico (SOP), también denominado hiperandrogenismo ovárico funcional o anovulación crónica hiperandrogénica, es una disfunción endocrino-metabólica de alta prevalencia. Es la causa más común de hiperandrogenismo con una incidencia de 3%, tanto en mujeres adolescentes como en adultas. Se estima que está presente en el 75% de las mujeres hirsutas y en el 10% de las mujeres premenopaúsicas. En los últimos años se ha podido establecer que este trastorno no sólo está limitado a la mujer en etapa reproductiva, sino que puede manifestarse desde el período prepuberal y, quizás, desde antes.
Además de la disfunción ovulatoria y del hiperandrogenismo que son las características más relevantes del síndrome, la mayoría de las mujeres con SOP (60-80%) presentan resistencia insulínica (RI) periférica, la que afecta principalmente al músculo y al tejido adiposo, y una hiperinsulinemia compensatoria que puede manifestarse en forma independiente de la obesidad1. La RI, en conjunto con la disfunción de la célula beta pancreática, constituye una comorbilidad frecuente en estas pacientes y juega un papel preponderante en las consecuencias metabólicas a largo plazo del síndrome, entre las que cabe destacar la diabetes tipo 2, la enfermedad cardiovascular y el hígado graso no alcohólico2,3. De hecho, más del 40% de estas pacientes desarrolla intolerancia a la glucosa y el 16% de ellas diabetes tipo 2 al final de la cuarta década de la vida. Más aún, se ha demostrado que la enfermedad coronaria es más frecuente en mujeres con SOP y que el riesgo de presentar infarto al miocardio es siete veces superior4. Todo esto ha sido de tal trascendencia que llevó a Reaven a incorporar al SOP dentro del síndrome X o plurimetabólico de Reaven junto con sus otros componentes (diabetes tipo 2, hipertensión arterial, dislipidemia, etc.), con lo cual el SOP, descrito inicialmente como un trastorno netamente reproductivo, quedó definido como un trastorno fundamentalmente endocrino-metabólico. Por lo tanto, es evidente, según todos estos argumentos, que el SOP debe ser considerado como un problema de salud pública que afecta a la mujer más allá de su esfera reproductiva pero que, no obstante, la lleva a consultar a edades tempranas y ofrece justamente la oportunidad de detectar en forma precoz las anormalidades metabólicas asociadas.
Si bien este síndrome fue descrito hace más de siete décadas, sigue siendo un tema de gran controversia e interés debido a su heterogeneidad, compleja fisiopatología y a los riesgos reproductivos y metabólicos que involucra. En el presente artículo revisaremos sólo algunos aspectos del síndrome que, a juicio y según la experiencia del autor, son los más novedosos y relevantes.
Diagnóstico del SOP y nuevos fenotipos
En 19355, Stein y Leventhal describieron una entidad clínica consistente en trastornos menstruales, esterilidad, hirsutismo y obesidad. Además, los ovarios de esas pacientes presentaban ciertas características morfológicas particulares tales como: aumento de tamaño, engrosamiento de la túnica albugínea y microquistes múltiples situados periféricamente en la zona subcortical ovárica. Posteriormente, en 1965, Smith y cols6 en un estudio de 301 casos, pusieron de manifiesto que los límites de esta entidad no eran tan precisos. De acuerdo a este estudio, el 40% de los casos tenía ovarios de tamaño normal y un 46% no presentaba engrosamiento de la túnica albugínea. Una investigación posterior demostró que el síndrome clínico podía asociarse a ovarios de morfología aparentemente normal y otro estudio estableció que entre el 16% y el 25% de las mujeres sanas podía presentar imágenes ultrasonográficas sugerentes de ovarios poliquísticos sin el síndrome clínico7, todo lo cual indicaría que el clásico síndrome de Stein Leventhal sería una excepción. Con el fin de unificar criterios y dada la heterogeneidad del síndrome, se lo definió en 1990 en una conferencia de consenso de la National Institutes of Health de EE.UU., como la “presencia de hiperandrogenismo asociado a anovulación crónica, sin otra causa específica de enfermedad suprarrenal o hipofisiaria que curse con irregularidades menstruales o exceso de andrógenos”8. Esta definición, evidentemente trató de dar a este síndrome un límite y un carácter de unidad, sin embargo, tenía la desventaja de englobar bajo un mismo concepto una serie de entidades diferentes. Además, no se consideró en esta definición el aspecto morfológico de los ovarios. Posteriormente, la Sociedad Europea de Reproducción y Embriología (ESHRE) y la Sociedad Americana de Medicina Reproductiva (ASRM), en una conferencia de consenso realizada en Rotterdam en el año 2003, propuso una nueva definición del síndrome que incorporó la presencia ecográfica de ovarios poliquísticos como un criterio diagnóstico9. Se propuso que luego de excluir otras formas de hiperandrogenismos, el SOP podía ser diagnosticado en pacientes que presentaran a lo menos dos de las tres características siguientes: hiperandrogenismo clínico o bioquímico; oligo-ovulación y presencia de ovarios de morfología poliquística, dando origen a cuatro fenotipos (Tabla1). De estos, los fenotipos A y B cumplen con los criterios NIH y son los que podríamos considerar un “SOP clásico”. Mientras que lo fenotipos C (hiperandrogenismo más ovarios poliquísticos, pero con ciclos ovulatorios, “SOP ovulatorio”) y D (oligo-ovulación más ovarios poliquísticos, pero sin evidencias clínicas o bioquímicas de hiperandrogenismo) son otras entidades que no cumplen con los criterios NIH y que han suscitado nuevas controversias10,11, sobretodo el fenotipo D, que por no tener hiperandrogenismo ni hiperandrogenemia, para algunos expertos no debería incluirse dentro del espectro del SOP, asumiendo que, por definición, el SOP es un trastorno eminentemente hiperandrogénico12. No obstante, a juicio del autor se requiere de más estudios y de seguimiento de los casos para poder incorporar o excluir definitivamente estos dos últimos fenotipos que son los más polémicos. Más aún, según nuestra experiencia cuando hemos realizado estudios familiares de SOP observamos que las hermanas del caso índice presentan estos fenotipos C y D, los que podrían representar casos atenuados de SOP dentro de un amplio espectro de manifestaciones clínicas. Este concepto ha sido recientemente corroborado por el grupo de Franks y col13. Otro aspecto interesante de hacer notar es que los fenotipos A y B que corresponden a la forma clásica de SOP son aparentemente los que presentan más RI y, por ende, más riesgo de enfermedades metabólicas que los fenotipos C y D14. En la Figura 1 se muestra la distribución de los distintos fenotipos en doscientas mujeres chilenas del área occidente de Santiago en las cuales se hizo el diagnóstico de SOP según los criterios de Rotterdam. Como se desprende de la figura, el fenotipo más prevalente fue el A seguido por el fenotipo B que corresponden a las formas clásicas de SOP. Es probable que en este estudio preliminar los otros fenotipos se encuentren subdimensionados debido a que, por ser menos sintomáticos, consulten en centros no especializados.
Después de 60 años aún no hay consenso en el diagnóstico de este síndrome que es altamente prevalente. En caso de duda hay que tener presente que el SOP es de mayor prevalencia aún en ciertos grupos de riesgo tales como pacientes con: BMI elevado; IR; diabetes mellitus 1 y 2; hirsutismo; oligo-ovulación; ovarios poliquísticos en la ecografía; pubarquia prematura; diabetes gestacional y retardo del crecimiento intrauterino15,20. Además, se debe tener especial cuidado con las adolescentes en las cuales es particularmente difícil establecer el diagnóstico de SOP debido a los cambios fisiológicos del eje somatotrópico y reproductivo propio de las niñas durante esta etapa de su desarrollo sexual. En ausencia de los factores de riesgo anteriormente mencionados, se sugiere controlar por un tiempo prudente, después de establecida la menarquia, para evitar el sobrediagnóstico21.
Etiopatogenia
Debido a la alta prevalencia de mujeres afectadas se postuló hace ya varios años que este síndrome podría tener una base genética. Lo anterior ha sido evaluado en diferentes poblaciones mediante estudios fenotípicos y de agregación familiar22. La mayoría de estas investigaciones han considerado el hiperandrogenismo y la morfología ovárica, estimada por ecografía y han establecido una alta frecuencia de SOP en las madres y hermanas de las pacientes23. Un escaso número de trabajos ha evaluado el compromiso metabólico en otros miembros de la familia de mujeres con SOP; uno de ellos sugiere que los familiares de primer grado de las pacientes con SOP presentarían niveles elevados de insulina24 y otro que la disfunción de la célula beta en familias de mujeres con SOP sería un rasgo hereditario25. Según nuestra experiencia, en un primer estudio de agregación familiar se logró establecer que la probabilidad de que un miembro de la familia (hermanos, padres y abuelos) de una mujer con SOP presentara diabetes tipo 2 era significativamente mayor que el de una mujer control26. Posteriormente demostramos que los padres de mujeres con SOP presentaban RI y diabetes tipo 2 con mayor frecuencia que los padres de mujeres normales, por lo que constituirían un grupo de alto riesgo de desarrollar diabetes tipo 227. De este estudio se desprende que el SOP podría ser considerado un marcador de una patología familiar, debido a que identifica riesgos metabólicos y, por consiguiente, el diagnóstico oportuno de SOP no sólo permitiría instaurar medidas terapéuticas y preventivas en los casos afectados sino que también en el grupo familiar. Otro aspecto destacable de este trabajo fue que los padres de las mujeres con SOP presentaban alteraciones metabólicas más frecuentemente que las madres. Algunos estudios previos ya habían sugerido que la herencia del SOP podría ser fundamentalmente paterna, basado en la proporción de afectadas en las madres y hermanas de mujeres con SOP, que si bien es elevada no es absoluta. Hasta el momento no se ha podido establecer la forma de herencia, lo que radica fundamentalmente en la heterogeneidad del síndrome y la ausencia del fenotipo masculino. Se podría especular que si el SOP tiene una base genética, el varón debería portar el o los genes de susceptibilidad, al igual que la mujer. Algunos estudios sugieren que sería el hombre con recesos temporales prematuros el “fenotipo masculino de SOP”28 lo que no ha sido corroborado en nuestros estudios en hijos y hermanos adultos de mujeres con SOP29,31. Esto ha llevado a sugerir que el fenotipo masculino del SOP se relacionaría más bien con el componente metabólico del síndrome32.
En los aspectos genéticos del SOP destacan a lo menos tres alteraciones que permitirían identificar “genes candidatos” y regiones de susceptibilidad: anormalidades del metabolismo, de la producción de andrógenos y de la foliculogénesis. Si bien se han propuesto algunos “genes candidatos” para todas ellas, hasta la fecha, no se han identificado plenamente la o las alteraciones genéticas asociadas a este síndrome33,35, por lo que se considera al SOP como una enfermedad poligénica compleja, sujeta a influencias ambientales las cuales jugarían un papel fundamental en la expresión del fenotipo reproductivo y metabólico del síndrome22.
Es poco probable que el SOP pueda ser explicado sobre la base de un solo desorden genético, a pesar de que en una determinada familia un gen podría tener un efecto predominante. La identificación de marcadores genéticos permitiría identificar y prevenir el desarrollo de diabetes tipo 2 así como sus complicaciones, en las mujeres con SOP y en los miembros de sus familias. Debido a la alta prevalencia de diabetes tipo 2 en las mujeres con este síndrome y sus familiares, el SOP debería considerarse un marcador de una patología familiar, un camino a la diabetes y un problema de salud pública.
Por lo tanto, las evidencias indican que el SOP tendría una predisposición genética que podría ponerse de manifiesto aún antes de la menarquia36,38. Además, se ha propuesto que factores ambientales, ya sea durante la vida prenatal39 o postnatal15, llevarían a una expresión clínica y bioquímica del síndrome en la vida adulta. A este respecto, surge un nuevo concepto muy en boga y de gran relevancia que vincula la etiopatogenia del SOP con los sucesos que se producen durante la vida prenatal.
Ambiente intrauterino, un posible mecanismo etiológico en el desarrollo del SOP
Entre los sucesos deletéreos que se producen durante la vida intrauterina, cabe mencionar al retardo del crecimiento intrauterino, el que dará origen a niños pequeños para la edad gestacional (PEG) y la exposición prenatal a andrógenos (EPA), los cuales han sido relacionados con la etiopatogenia del SOP40. Se ha planteado que los niños PEG tienen mayor riesgo de desarrollar síndrome metabólico en la vida adulta y se ha sugerido, además, una relación entre el bajo peso de nacimiento y el desarrollo posterior de SOP41. A este respecto, hemos podido establecer que el antecedente de PEG es mayor en mujeres con SOP y que la prevalencia de PEG es significativamente mayor en los niños nacidos de madres con SOP26,42. Respecto al segundo determinante prenatal, se ha planteado que la EPA estaría relacionada con la etiopatogenia del SOP. Estudios en modelos animales (monas y ovejas) establecen que las hembras androgenizadas por una exposición prenatal a andrógenos durante la vida intrauterina, como consecuencia de la administración exógena de testosterona a las madres, desarrollan rasgos postpuberales típicos de SOP43 En humanos se ha observado que mujeres con deficiencia de la 21-hidroxilasa se autoandrogenizan durante la vida fetal desarrollando un SOP secundario en la vida postnatal. Estas observaciones sugieren que la EPA de origen materno y/o fetal sería un posible mecanismo etiopatogénico para el desarrollo de SOP. En el modelo animal es la madre la que traspasa el exceso de andrógenos al feto mientras que en el modelo humano (deficiencia de la 21-hidroxilasa) es la suprarrenal fetal la que produce el exceso de andrógenos generando una “autoandrogenización” y un SOP secundario. Estos resultados sugieren que la exposición prenatal a andrógenos podría explicar mucho de los rasgos típicos del SOP. En este sentido, cobran gran interés las observaciones efectuadas por nuestro grupo en mujeres con SOP y con amenorrea de la lactancia las cuales presentaban ovarios aumentados de volumen y una mayor concentración de androstenediona que las nodrizas normales44. Esto permitió inicialmente plantear que los ovarios de estas mujeres permanecieron esteroidogénicamente activos durante el embarazo, manteniendo una secreción androgénica elevada la que, eventualmente, podría haber androgenizado al feto. Posteriormente establecimos que las embarazadas con SOP efectivamente presentan niveles elevados de andrógenos, pudiendo constituir una fuente de exceso de andrógenos para el feto asemejándose al modelo experimental39.
Además de lo anteriormente expuesto debemos recordar que el feto femenino o masculino de una madre SOP se va a desarrollar en un ambiente materno hiperinsulinémico39,45, lo cual también podría influir en la concentración de esteroides46,47 (Figura 2).
En síntesis, podemos decir que, de acuerdo a nuestros estudios, la embarazada con SOP presenta elevadas concentraciones de andrógenos, mayores concentraciones de insulina y triglicéridos, menores concentraciones de adiponectina y una exacerbación de la RI fisiológica del embarazo, hechos que podrían actuar como factores de reprogramación fetal39,45,48.
Basado en estas observaciones, en los últimos diez años nuestro quehacer científico ha estado enfocado en estudiar la función reproductiva y metabólica de hijas e hijos de mujeres portadoras de un SOP clásico (fenotipo A) durante diferentes etapas del desarrollo sexual. Desde el punto de vista reproductivo, hemos demostrado que tanto las hijas como los hijos nacidos de madres con SOP presentan elevadas concentraciones de hormona anti-Mülleriana (AMH) durante el período prepuberal (infancia temprana y niñez), lo que sugiere una alteración del desarrollo folicular en las niñas y de la función Sertoli en los niños30,36,37. Debido a que las células de la granulosa y las de Sertoli tienen un origen embriológico común, se podría especular que factores genéticos y/o ambientales afectarían una línea celular específica durante el desarrollo de la gónada fetal. Otra observación interesante que hemos establecido es que las alteraciones metabólicas preceden al hiperandrogenismo en las hijas de madres con SOP y que los rasgos bioquímicos del SOP debutan en la etapa Tanner IV del desarrollo puberal con aumento de la testosterona y una respuesta aumentada de LH al luprón49. Desde el punto de vista metabólico, las hijas de madres con SOP muestran bajas concentraciones de adiponectina y aumento de las concentraciones de insulina y triglicéridos38, mientras que los hijos presentan sobrepeso desde la infancia y desarrollan RI a medida que se hacen más adultos31. Estos datos en conjunto sugieren que los hijos/as de madres con SOP constituyen un grupo de alto riesgo para desarrollar alteraciones metabólicas y reproductivas a lo largo de la vida.
En síntesis, nuestros resultados muestran que existiría un probable fenómeno de reprogramación fetal de los hijos de madres con SOP, que se evidencia tanto por una mayor frecuencia de niños PEG como por cambios en la función reproductiva y metabólica de estos niños. Debido a que en humanos es difícil disecar el aspecto genético del ambiental, los modelos experimentales de androgenización prenatal son de gran relevancia. En este sentido, el Dr. Recabarren, del Laboratorio de Fisiología y Endocrinología Animal de la Universidad de Concepción, ha desarrollado un modelo en ovejas de androgenización prenatal lo cual ha sido de gran utilidad para el estudio de la etiopatogenia del síndrome y nos ha permitido corroborar varias de las observaciones efectuadas en los hijos de estas pacientes50,52.
Enfoque terapéutico
El tratamiento debe estar orientado a corregir el hiperandrogenismo, la anovulación crónica y las alteraciones metabólicas asociadas a la RI y al hiperinsulinismo. La edad y el deseo o no de embarazo son factores decisivos en la elección inicial de la terapia, no obstante, la corrección de las alteraciones metabólicas debe preceder o acompañar a cualquier medida terapéutica. Por ser el SOP una disfunción endocrino-metabólica crónica con un fuerte componente genético, su curación espontánea es dudosa, por lo que los tratamientos deben iniciarse precozmente y ser prolongados. Un análisis detallado del tratamiento de cada uno de estos aspectos escapa al objetivo de esta revisión; quizás lo más importante es hacer énfasis en aquellas conductas y medidas terapéuticas que pudieran tener un potencial efecto en retardar o prevenir los riesgos a los que están expuestas estas pacientes. En primer lugar, debe combatirse la obesidad mediante dieta y ejercicio físico regular. La obesidad empeora la RI, las alteraciones metabólicas derivadas de la hiperinsulinemia y también agrava aspectos reproductivos del síndrome, como hiperandrogenismo, anovulación crónica, complicaciones obstétricas y mayor incidencia de cánceres dependientes de hormonas. Su tratamiento disminuye la hiperinsulinemia y puede atenuar las alteraciones metabólicas asociadas; disminuye los niveles de testosterona y LH permitiendo la reanudación espontánea de la ciclicidad ovárica y de la ovulación, o al menos aumenta la sensibilidad a los inductores de ovulación. Los anticonceptivos orales con progestinas de baja actividad androgénica no sólo son de gran utilidad en el manejo del hiperandrogenismo sino que también permiten una descamación regular del endometrio con lo que se evita el riesgo de hiperplasia endometrial y cáncer de endometrio y, por su efecto inhibitorio del eje ovárico, el desarrollo de cáncer ovárico. Por último, el nuevo concepto de la implicancia de la hiperinsulinemia en la patogenia del SOP, ha motivado el uso de drogas como la metformina que favorecen la señalización insulinica53. Hasta la fecha existen numerosos estudios clínicos con metformina en mujeres en edad reproductiva y en adolescentes con SOP54,57. Esta droga disminuye los niveles de insulina circulante; aumenta la concentración de SHBG; reduce la concentración de andrógenos y LH circulantes, lo que tendría un efecto beneficioso sobre el hirsutismo; disminuye el apetito, el peso corporal y el IMC y reanuda la ciclicidad ovárica y la ovulación. En las pacientes con RI que desean fertilidad, ya sean obesas o normales en peso, se recomienda usar metformina en una dosis promedio de 1500 mg/día. En un plazo de 4-6 meses se recupera la ciclicidad ovárica y aumenta la tasa de ovulación48. Debido a la alta prevalencia de obesidad y RI, las mujeres con SOP tienen un alto riesgo de presentar patologías del embarazo tales como diabetes gestacional e hipertensión del embarazo y una mala historia obstétrica con mayor incidencia de abortos del primer trimestre, partos prematuros, cesáreas y el nacimiento de un niño PEG o macrosómico42,48,58. Basado en estas observaciones se ha iniciado el uso de metformina durante el embarazo lo que tendría tres potenciales aplicaciones terapéuticas: disminuir el riesgo de abortos prematuros, el de patologías del embarazo y del período post-parto y el de androgenización prenatal59,61. Esta medida terapéutica aparentemente no tendría un efecto deletéreo sobre los hijos62.
Reflexiones y direcciones futuras
Por ser el SOP una disfunción endocrino metabólica con un variado espectro de anormalidades que se presentan a lo largo de toda la vida de la mujer, las pacientes suelen consultar en diversas especialidades donde, por lo general, se da énfasis al motivo de consulta con lo cual el diagnóstico de SOP pasa desapercibido. Lo anterior implica que estas pacientes deben ser diagnosticadas y tratadas oportunamente, necesitan ser informadas y educadas respecto a su patología y, finalmente, ser controladas en forma prolongada.
Las pacientes con SOP y sus familiares constituyen una población de alto riesgo para el desarrollo de diabetes tipo 2. Por ello, mirado no solo desde un punto de vista individual sino que desde un contexto epidemiológico, el diagnóstico de SOP es extremadamente importante.
El futuro de este síndrome estará en la identificación de fenotipos y marcadores genéticos que permitan instaurar medidas preventivas precoces y en el manejo adecuado de las embarazadas con SOP, con el fin de reducir el desarrollo de alteraciones metabólicas y reproductivas en los hijos y la perpetuación de este síndrome.

X corresponde al criterio; + corresponde a la clasificación.
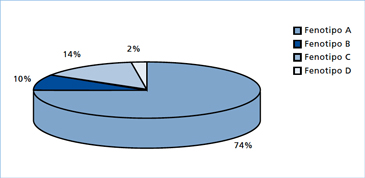
Figura 1. Frecuencia de los distintos fenotipos de SOP según los criterios de Rotterdam en una población de mujeres del área Occidente de Santiago.
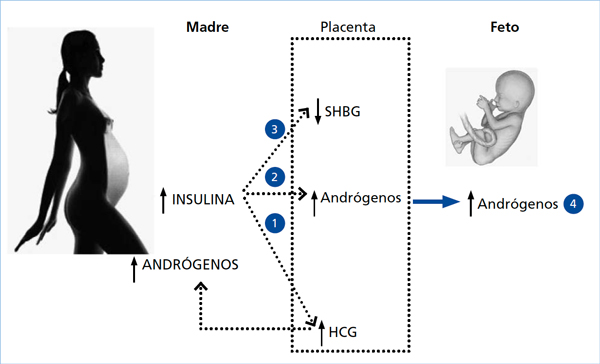
Figura 2. Modelo de androgenización prenatal en mujeres con SOP durante el embarazo.
(1) Los elevados niveles de insulina podrían inducir un aumento de la concentración de HCG lo cual estimularía al ovario materno y/o a la gónada fetal, aumentando los niveles de andrógenos en ambos compartimientos; (2) A su vez, la insulina podría aumentar la actividad de las enzimas 3ß-hidroxiesteroide deshidrogenasa tipo 1 y/o disminuir la actividad P450arom llevando a una mayor síntesis de andrógenos por la placenta y (3) la hiperinsulinemia materna podría disminuir la expresión de SHBG placentaria con lo cual aumentaría la concentración de andrógenos libres. (4) Estos tres mecanismos por sí solos o en forma sinérgica llevarían a un aumento de los andrógenos en el compartimiento fetal.
Referencias
- Dunaif A., Segal KR., Futterweit W., Dobrjansky A. 1989. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 38:1165–1174.
- Legro RS., Kunselman AR., Dodson WC., Dunaif A. 1999. Prevalence and predictors of risk for Type II diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab 84:165–169.
- Cerda C., Pérez-Ayuso RM., Riquelme A., Soza A., Villaseca P., Sir- Petermann T., Espinoza M., Pizarro M., Solis N., Miquel JF., Arrese M. 2007. Nonalcoholic fatty liver disease in women with polycystic ovary syndrome. J Hepatol. 47:412-417.
- Dahlgren E., Janson PO., Johansson S., Lapidus L., Oden A. 1992. Polycystic ovary syndrome and risk for miocardial infarction. Evaluated from a risk factor model based on prospective population study of women. Acta Obstet Gynecol Scand 71:599-603.
- Stein IF., Leventhal ML. 1935. Amenorrhea associated with bilateral polycystic ovaries. Am J Obstet Gynecol 29: 181-191.
- Smith KD., Steinberger E., Perloff WH. 1965. Polycystic ovarian disease (PCO). A report of 301 cases. Am J Obstet Gynecol 93:994-1001.
- Polson DW., Adams J., Wadsworth J., Franks S. 1988. Polycystic ovaries: a common finding in normal woman. Lancet 1:870-878.
- Zawadski JK., Dunaif A. 1992. Diagnostic criteria for polycystic ovary syndrome: towards a rationale approach. En: Polycystic Ovary Syndrome, ed. por Dunaif A, Givens J R, Haseltine F, Merriam G R, Boston: Blackwell 377-384.
- Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group 2004. Revised 2003 consensus on diagnostic criteria and longterm health risks related to polycystic ovary syndrome. Fertil Steril. 81:19-25.
- Franks S. 2006. Controversy in clinical endocrinology: diagnosis of polycystic ovarian syndrome: in defense of the Rotterdam criteria. J Clin Endocrinol Metab. 91:786-789.
- Azziz R. 2006. Controversy in clinical endocrinology: diagnosis of polycystic ovarian syndrome: the Rotterdam criteria are premature. J Clin Endocrinol Metab. 91:781-785
- Azziz R., Carmina E., Dewailly D., Diamanti-Kandarakis E., Escobar- Morreale HF., Futterweit W., Janssen OE., Legro RS., Norman RJ., Taylor AE., Witchel SF. 2006. Androgen Excess Society. Positions statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab. 91:4237-4245.
- Franks S., Webber LJ., Goh M., Valentine A., White DM., Conway GS., Wiltshire S., McCarthy MI. 2008. Ovarian morphology is a marker of heritable biochemical traits in sisters with polycystic ovaries. J Clin Endocrinol Metab (en prensa).
- Chang WY., Knochenhauer ES., Bartolucci AA., Azziz R. 2005. Phenotypic spectrum of polycystic ovary syndrome: clinical and biochemical characterization of the three major clinical subgroups. Fertil Steril. 83:1717-1723.
- Gambineri A., Pelusi C., Vicennati V., Pagotto U., Pasquali R. 2002. Obesity and the polycystic ovary syndrome. Int J Obes Relat Metab Disord. 26: 883-896.
- Codner E., Escobar-Morreale HF. 2007. Clinical review: Hyperandrogenism and polycystic ovary syndrome in women with type 1 diabetes mellitus. J Clin Endocrinol Metab. 92:1209-1216.
- Mericq V., Ong KK., Bazaes R., Peña V., Avila A., Salazar T., Soto N., Iñiguez G., Dunger DB. 2005. Longitudinal changes in insulin sensitivity and secretion from birth to age three years in small- and appropriate- for-gestational-age children. Diabetologia. 48:2609-2614.
- . Ibáñez L., Potau N., Zampolli M., Street ME., Carrascosa A. 1997. Girls diagnosed with premature pubarche show an exaggerated ovarian androgen synthesis from the early stages of puberty: evidence from gonadotropin- releasing hormone agonist testing. Fertil Steril. 67:849-855.
- Lesser KB., Garcia FA. 1997. Association between polycystic ovary syndrome and glucose intolerance during pregnancy. J Matern Fetal Med. 6:303-307.
- Ibáñez L., Potau N., Francois I., de Zegher F. 1998. Precocious pubarche, hyperinsulinism, and ovarian hyperandrogenism in girls: relation to reduced fetal growth. J Clin Endocrinol Metab. 83:3558-3562.
- Witchel SF. 2006. Puberty and polycystic ovary syndrome. Mol Cell Endocrinol. 254-255:146-153.
- Escobar-Morreale HF., Luque-Ramírez M., San Millán JL. 2005. The molecular-genetic basis of functional hyperandrogenism and the polycystic ovary syndrome. Endocr Rev. 26:251-282.
- Battaglia C., Regnani G., Mancini F., Iughetti L., Flamigni C., Venturoli S. 2002. Polycystic ovaries in childhood: a common finding in daughters of PCOS patients. A pilot study. Hum Reprod. 17:771-776.
- Norman RJ., Masters S., Hague W. 1996. Hyperinsulinemia is common in family members of women with polycystic ovary syndrome. Fertil Steril 66:942-947.
- Colilla S., Cox NJ., Ehrmann DA. 2001. Heritability of insulin secretion and insulin action in women with polycystic ovary syndrome and their first degree relatives. J Clin Endocrinol Metab. 86:2027-2031.
- Benítez R., Sir-Petermann T., Palomino A., Angel B., Maliqueo M., Pérez F., Calvillán M. 2001. Prevalencia familiar de patologías metabólicas en pacientes con síndrome de ovario poliquístico. Rev Med Chil 129:707-712.
- Sir-Petermann T., Angel B., Maliqueo M., Carvajal F., Santos JL., Pérez-Bravo F. 2002. Prevalence of Type II diabetes mellitus and insulin resistance in parents of women with polycystic ovary syndrome. Diabetologia 45:959-964.
- Govind A., Obhrai MS., Clayton RN. 1999.Polycystic ovaries are inherited as an autosomal dominant trait: analysis of 29 polycystic ovary syndrome and 10 control families. J Clin Endocrinol Metab 84:38-43.
- Sir-Petermann T., Cartes A., Maliqueo M., Vantman D., Gutiérrez C., Toloza H., Echiburú B., Recabarren SE. 2004. Patterns of hormonal response to the GnRH agonist leuprolide in brothers of women with polycystic ovary syndrome: a pilot study. Hum Reprod. 19:2742-2747.
- Recabarren SE., Sir-Petermann T., Rios R., Maliqueo M., Echiburú B., Smith R., Rojas-García P., Recabarren M., Rey RA. 2008 Pituitary and testicular function in sons of women with polycystic ovary syndrome from infancy to adulthood. J Clin Endocrinol Metab. (en prensa).
- Recabarren SE., Smith R., Rios R., Maliqueo M., Echiburú B., Codner E., Cassorla F., Rojas P., Sir-Petermann T. 2008. Metabolic profile in sons of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 93:1820-1826.
- Azziz R. 2008. Polycystic ovary syndrome is a family affair. J Clin Endocrinol Metab. 93:1579-1581.
- Sir-Petermann T., Pérez-Bravo F., Angel B., Maliqueo M., Calvillan M., Palomino A. 2001 G972R polymorphism of IRS-1 in women with polycystic ovary syndrome. Diabetologia. 44:1200-1251.
- Sir-Petermann T., Angel B., Maliqueo M., Santos JL., Riesco MV., Toloza H., Pérez-Bravo F. 2004. Insulin secretion in women who have polycystic ovary syndrome and carry the Gly972Arg variant of insulin receptor substrate-1 in response to a high-glycemic or low-glycemic carbohydrate load. Nutrition. 20:905-910.
- Pérez-Bravo F., Echiburú B., Maliqueo M., Santos JL., Sir-Petermann T. 2005. Tryptophan 64 --> arginine polymorphism of beta-3-adrenergic receptor in Chilean women with polycystic ovary syndrome. Clin Endocrinol (Oxf). 62:126-131.
- Sir-Petermann T., Codner E., Maliqueo M., Echiburú B., Hitschfeld C., Crisosto N., Pérez-Bravo F., Recabarren SE., Cassorla F. 2006. Increased anti-Müllerian hormone serum concentrations in prepubertal daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 91:3105-3109.
- Crisosto N., Codner E., Maliqueo M., Echiburú B., Sánchez F., Cassorla F., Sir-Petermann T. 2007. Anti-Müllerian hormone levels in peripubertal daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 92:2739-2743.
- Sir-Petermann T., Maliqueo M., Codner E., Echiburú B., Crisosto N., Pérez V., Pérez-Bravo F., Cassorla F. 2007. Early metabolic derangements in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 92:4637-4642.
- Sir-Petermann T., Maliqueo M., Angel B., Lara HE., Perez-Bravo F., Recabarren SE. 2002 Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod 17:2573-2579.
- Xita N., Tsatsoulis A. 2006. Fetal Programming of Polycystic Ovary Syndrome by Androgen Excess: Evidence from Experimental, Clinical and Genetic Association Studies. J Clin Endocrinol Metab. 91:1660- 1666.
- Sir-Petermann T., Hitschfeld C., Codner E., Maliqueo M., Avila A., Echiburú B., Gacitúa R., Crisosto N., Cassorla F. 2007. Gonadal function in low birth weight infants. J Pediatr Endocrinol Metab. 20:405- 414.
- Sir-Petermann T., Hitchsfeld C., Maliqueo M., Codner E., Echiburú B., Gacitúa R., Recabarren SE., Cassorla F. 2005. Birth weight in offspring of mothers with polycystic ovary syndrome. Hum. Reprod. 20:2122- 2126.
- Padmanabhan V., Manikkam M., Recabarren S., Foster D. 2006. Prenatal testosterone excess programs reproductive and metabolic dysfunction in the female. Mol Cell Endocrinol. 246:165-174.
- Sir-Petermann T., Devoto L., Maliqueo M., Peirano P., Recabarren SE., Wildt L. 2001 Resumption of ovarian function during lactational amenorrhoea in breastfeeding women with polycystic ovary syndrome: endocrine aspects. Hum Reprod 16:1603-1610.
- Sir-Petermann T., Echiburú B., Maliqueo MM., Crisosto N., Sánchez F., Hitschfeld C., Cárcamo M., Amigo P., Pérez-Bravo F. 2007. Serum adiponectin and lipid concentrations in pregnant women with polycystic ovary syndrome. Hum Reprod. 22:1830-1836.
- Nestler JE. 1989. Insulin and insulin-like growht factor I stimulate the 3ß-hydroxisteroid dehydrogenase activity of human placental cytotrophoblast. Endocrinology 125:2127-2133.
- Queipo G., Deas M., Arranz C., Carino C., Gonzalez R., Larrea F. 1998. Sex hormone-binding globulin stimulates chorionic gonadotrophin secretion from human cytotrophoblasts in culture. Hum Reprod. 13:1368-1373.
- Villarroel AC., Echiburú B., Riesco V., Maliqueo M., Cárcamo M., Hitschfeld C., Sánchez F., del Solar MP., Sir-Petermann T. 2007. Síndrome de ovario poliquístico (SOP) y embarazo: Experiencia clínica Rev Med Chil. 135:1530-1538.
- Sir-Petermann T., Perez V., Ladron de Guevara A., Diaz E., Maliqueo M., Echiburu B., Codner E., Cassorla F., Crisosto N. 2008. Metabolic and reproductive features in peri-pubertal daughters of women with polycystic ovary syndrome. The Endocrine Society’s 90th Annual Meeting, San Francisco, USA. [P3-577].
- Recabarren SE., Padmanabhan V., Codner E., Lobos A., Durán C., Vidal M., Foster DL., Sir-Petermann T. 2005. Postnatal developmental consequences of altered insulin sensitivity in female sheep treated prenatally with testosterone. Am J Physiol Endocrinol Metab. 289: E801-806.
- Recabarren SE., Lobos A., Figueroa Y., Padmanabhan V., Foster DL., Sir-Petermann T. 2007. Prenatal testosterone treatment alters LH and testosterone responsiveness to GnRH agonist in male sheep. Biol Res.40:329-338.
- Recabarren SE., Rojas-Garcia PP., Recabarren MP., Alfaro VH., Smith R., Padmanabhan V., Sir-Petermann T. 2008. Prenatal testosterone excess reduces sperm count and motility Endocrinology. (en prensa).
- Gunton JE., Delhanty JD., Takahashi S., Baxter RC. 2003. Metformin rapidly increases insulin receptor activation in human liver and signals preferentially through insulin-receptor substrate-2. J. Clin. Endocrinol. Metab. 88:1323-1332.
- De Leo V., la Marca A., Petraglia F. 2003. Insulin-lowering agents in the management of polycystic ovary syndrome. Endocr Rev. 24:633- 667.
- Moll E., van der Veen F., van Wely M. 2007. The role of metformin in polycystic ovary syndrome: a systematic review. Hum Reprod Update. 13:527-537.
- De Leo V., Musacchio MC., Morgante G., Piomboni P., Petraglia F. 2006. Metformin treatment is effective in obese teenage girls with PCOS. Hum Reprod. 21:2252-2256.
- Ibáñez L., de Zegher F. 2005. Flutamide-Metformin plus ethinylestradiol- drospirenone for lipolysis and antiatherogenesis in young women with ovarian hyperandrogenism: The key role of metformina at the start and after more than one year of therapy. J. Clin. Endocrinol. Metab. 90:39-43.
- Boomsma CM., Eijkemans MJ., Hughes EG., Visser GH., Fauser BC., Macklon NS. 2006. A meta-analysis of pregnancy outcomes in women with polycystic ovary syndrome. Hum Reprod Update. 12:673-683.
- Khattab S., Mohsen IA., Foutouh IA., Ramadan A., Moaz M., Al-Inany H. 2006. Metformin reduces abortion in pregnant women with polycystic ovary syndrome. Gynecol Endocrinol. 22:680-684.
- Glueck CJ., Goldenberg N., Wang P., Loftspring M., Sherman A. 2004. Metformin during pregnancy reduces insulin resistance, insulin secretion, weight, testosterone and development of gestational diabetes: prospective longitudinal assessment of women with polycystic ovary syndrome from preconception throughout pregnancy. Hum Reprod 19:510-521.
- Rowan JA., Hague WM., Gao W., Battin MR., Moore MP., MiG Trial Investigators 2008. Metformin versus insulin for the treatment of gestational diabetes. N Engl J Med. 358:2003-2015.
- Glueck CJ., Goldberg N., Pranikoff J., Loftspring M., Sieve L., Wang P. 2004. Height, weight, and motor social development during the first 18 months of life in 126 infants born to 109 mothers with polycystic ovary syndrome who conceived on and continued metformina through pregnancy. Hum Reprod 19:1323-1330.
Agradecimientos
El autor agradece a sus colegas y colaboradores, Sr. Manuel Maliqueo y Srta. Bárbara Echiburú, y al personal del laboratorio, Sra. Estela González y Sra. Evelyn Velásquez, quienes han hecho posible el desarrollo de esta línea de investigación. Estos estudios han sido financiados por el Fondo Nacional de Desarrollo Científico y Tecnológico y la Fundación Alexander Von Humboldt.