Uso de hormona de crecimiento en la transición desde el adolescente a adulto
Alejandro Martínez-Aguayo1, Carlos Torres B.2 y Carmen Carrasco M.3
Growth hormone use in the transition phase from adolescence to adulthood
1Unidad de Endocrinología, División de Pediatría,
Facultad de Medicina, Pontificia Universidad Católica de
Chile, Santiago, Chile.
2Unidad de Endocrinología, Servicio de Pediatría,
Hospital Guillermo Grant Benavente, Concepción, Chile
3Departamento de Endocrinología, Facultad de
Medicina, Pontificia Universidad Católica de Chile,
Santiago, Chile.
Correspondencia:
Dr. Alejandro Martínez- Aguayo
División de Pediatría, Escuela de Medicina
Pontificia Universidad Católica de Chile
Lira 85, piso 5, Santiago, Chile.
Código postal: 8330074
Teléfono: (56-2) 354-3095 Fax: (56-2) 638-5675
E-mail: alemarti@med.puc.cl
Recibido: 14 de Junio de 2012
Aceptado: 19 de Junio de 2012
The transition phase is defined as the period in which linear growth ends and an optimal muscle and bone mass is achieved. An important proportion of individuals with isolated growth hormone deficiency, do not require growth hormone supplementation during adulthood, differing from subjects with multiple pituitary hormone deficiency. Growth hormone secretion must be reassessed in patients with isolated deficiency, when linear growth ends. The ideal is to perform an insulin tolerance test, which is considered adequate when a growth hormone peak over 6.1 ng/ml is achieved. Subjects with multiple pituitary hormone deficiency do not require this test if their IGF-1 value is below 2 standard deviations of normal. An initial dose of 12.5 ug/kg/day is recommended, that must be titered to maintain IGF-1 levels in the upper quartile of normal. Patients who require growth hormone, are benefited with a higher bone mineral and muscle mass and a better quality of life.
Key words: Growth hormone, phase transition, deficiency of growth hormone isolated
syndrome, growth hormone deficiency in adulthood multiple pituitary hormone deficiency.
El manejo óptimo de los adolescentes con deficiencia de hormona de crecimiento (DHC) continúa siendo un desafío tanto para los endocrinólogos pediátricos como de adultos. El tratamiento con hormona de crecimiento (HC) en pacientes adultos fue aprobado en Agosto 1996 en los Estados Unidos. Uno de los principales objetivos del tratamiento en los pacientes con DHC es lograr una talla final lo más cercana a la talla objetivo genética. Pero existen otros objetivos que están relacionados con lograr un desarrollo óptimo de la masa ósea, composición corporal, fuerza muscular y calidad de vida1-4.
Se ha considerado que la talla final de un individuo se logra en los varones cuando alcanzan una edad ósea entre los 16,5 a 17 años y en las mujeres una edad de 14,5 a 15 años. En cambio el peak de masa ósea y de masa muscular se logran un poco más tarde, después de los 20 años en ambos sexos. Aproximadamente un 37% de la masa ósea de logra durante la pubertad hasta los 20 a 35 años de edad5.
Se define Fase de Transición, el período de tiempo que transcurre entre el término del crecimiento lineal (menor a 2 cm por año) y el desarrollo óptimo muscular y de densidad mineral ósea. Durante este período de tiempo ocurren importantes cambios físicos y psicológicos, que se extienden usualmente por 6 a 7 años después de logrado la talla final3. La HC no sólo actúa estimulando el crecimiento lineal, sino que también posee importantes efectos en promover la lipólisis, masa muscular, densidad mineral ósea, adecuada función cardíaca, tolerancia al ejercicio y calidad de vida6,7.
La Fase de Transición es importante porque se debe establecer sí los adolescentes que han sido tratados por DHC aislada durante la infancia persisten con deficiencia en la vida adulta, ya que ellos requieren tratamiento prolongado y probablemente de por vida. Los pacientes adultos con DHC tienen un síndrome que se caracteriza por: una composición corporal metabólicamente adversa, menor densidad mineral ósea (con mayor riesgo de fractura que se exacerba cuando esta asociado a hipogonadismo o cuando ha ocurrido un sobre-tratamiento con glucocorticoides), menor capacidad de ejercicio, alteraciones en el metabolismo de carbohidratos y lípidos (incremento del colesterol LDL y triglicéridos con bajas concentraciones de colesterol HDL). Esto se traduce en una peor calidad de vida. El uso de HC en pacientes con DHC diagnosticado en la vida adulta es aún motivo de controversia; sin embargo, existe mayor consenso en la conducta a adoptar en los pacientes a los cuales el diagnóstico se hizo durante la niñez6.
Los efectos de la DHC severa se muestran en la Tabla 1. Estos síntomas y signos mejoran con el uso de HC en dosis relativamente bajas, comparadas a las necesarias durante la niñez3.
Tabla 1.

¿Quién, cuándo y cómo se debe
re-evaluar el diagnóstico de deficiencia
de hormona de crecimiento durante la
transición?
¿Qué sujetos requieren re-evaluación del diagnóstico de DHC?
Un número importante de niños que han sido diagnosticados con DHC, al ser re- evaluados durante la adolescencia o adultez no resultan deficientes. Esto ocurre especialmente en los casos de deficiencia aislada o “idiopática”. En promedio un 67% (rango; 40 a 75%) de los adolescentes con DHC aislada persisten deficientes en la vida adulta al ser reevaluados8-11. Esto sugiere la necesidad de repetir las pruebas de estimulo en los pacientes con DHC aislada o deficiencia idiopática de HC cuando terminan el crecimiento lineal.
Los pacientes con DHHM, o individuos con mutaciones conocidas en genes que controlan el desarrollo y/o expresión de HC, o sujetos con alteraciones estructurales de la región hipotálamo hipofisaria, tales como agenesia de cuerpo calloso, displasia septo-óptica, etc tienen una alta posibilidad de persistir con deficiencia de HC en la vida adulta (mayor que un 90%)12 y no requieren necesariamente de re-evaluación13,14. En cambio, un 50% de los pacientes que han tenido DHC por radiación durante la infancia, no cumple criterio de DHC en la vida adulta15; esto indica que estos sujetos sí deben ser re-evaluados. Sin embargo, las pruebas de estímulo de estos pacientes deben ser interpretados con cautela ya que pueden tener alteración en la neuro-secreción de HC y de esta forma responder a estímulos farmacológicos pero no en condiciones fisiológicas.
Hartman et al13, han sugerido que, en aquellos individuos con DHHM u otros con alta probabilidad de tener deficiencia de HC en la vida adulta, se suspenda la HC durante un mes una vez que hayan culminado el crecimiento lineal y se determinen las concentraciones de IGF-1. Si ésta es menor a -2 DE, no sería necesario una re-evaluación con pruebas de estimulo para HC y se podría continuar con HC en bajas dosis16,17.
¿Cuándo se debe realizar la re-evaluación?
La guía de la “GH Reseach Society” sugiere que, una vez suspendida la HC, es necesario esperar entre 1 a 3 meses antes de re-evaluar1,6. Los sujetos con DHHM deben continuar recibiendo las otras hormonas (cortisol, levotiroxina, esteroides sexuales, etc) en dosis que aseguren un remplazo adecuado.
Recientemente, un grupo italiano18 sugirió que aquellos pacientes con DHC aislada diagnosticados antes de la pubertad pueden ser re-evaluados en la mitad del desarrollo puberal. Estos autores observaron que un tercio de ellos tenían un peak de HC mayor a 10 ng/mL, en base a lo cual se suspendió el tratamiento de HC logrando una talla final adecuada. Sin embargo, este trabajo debe ser replicado antes que constituya una recomendación.
¿Cómo evaluar?
Aún es materia de controversia el punto de corte para definir deficiencia de HC al utilizar las pruebas de estímulo. Estos valores han sido definidos arbitrariamente para la fase de transición y algunos expertos sugieren un valor intermedio entre niños y adultos (5,0 ng/mL). Sin embargo, este valor no tiene un fundamento fisiológico y depende de la potencia del “secretagogo”, del índice de masa corporal y del tipo de ensayo bioquímico para determinar HC.
Se sabe que la producción de HC, más alta en la adolescencia, coincide con el peak de velocidad de crecimiento19 y persiste hasta la mitad de la década de los 20 años, cuando progresivamente comienza a disminuir con la edad. Se puede observar cierto grado de hiposomatotropismo en la vejez20. Considerando las diferencias fisiológicas entre adolescentes y adultos, es lógico que no deba usarse el mismo punto de corte para hacer el diagnóstico, como tampoco la misma dosis de HC.
La prueba de estímulo con insulina para inducir hipoglicemia (ITT, insulin tolerance test) es la mejor prueba para detectar deficiencias de HC en la vida adultos1,6,21 y debe ser la prueba de elección durante la fase de transición1,16,17. En adultos se considera normal un peak de > 3,0 ng/mL21, pero este punto de corte es muy restrictivo para los adolescentes en transición. Maghnie et al14, estudiaron la respuesta de HC medida por IRMA en ITT en controles sanos entre los 15 a 30 años. Estos investigadores sugieren un punto de corte de 6,1 ng/mL, el cual tiene un 96% de sensibilidad y 100% de especificidad para detectar deficiencia de HC en la fase de transición. Secco et al22, realizaron un estudio similar midiendo GH por ECLIA y concluyen que un punto de corte de 5,6 ng/ml tendría una sensibilidad de 77,4% y especificidad de 93,8%. El ITT sólo se puede realizar en un centro terciario con alta experiencia en esta prueba por los eventos adversos que puede ocurrir y está contraindicado en sujetos con antecedentes de convulsiones o con riesgo cardiovascular conocido. La prueba con clonidina no debe ser usada en esta etapa.
La prueba con GHRH-arginina es una alternativa razonable al ITT; sin embargo, los puntos de corte no son uniformes entre diferentes grupos y las concentraciones varían desde 1,5 a 5,0 ng/mL (IRMA). Un valor intermedio de 4 ng/mL permitiría alcanzar un 95% de sensibilidad y 92% de especificidad3,17.
En los sujetos con DHC aislada, las concentraciones de IGF-1 no deben ser consideradas como criterio diagnóstico en la re-evaluación. Maghnie et al14,23, plantean que una IGF- 1 menor a -1,7 DE es sugerente de DHC, pero hay que considerar que la IGF-1 puede no disminuir bajo lo normal hasta 6 meses posterior a haber descontinuado HC14. En los pacientes con DHHM, la determinación de IGF-1 es útil, aquellos sujetos con una concentración menor a -2 DE se define como DHC y no requieren prueba de estímulo.
Beneficio de usar GH en un paciente adulto con deficiencia de hormona de crecimiento
Composición corporal
La importancia de utilizar HC en los pacientes adultos con DHC severa es evitar un mayor riesgo cardiovascular Los beneficios de usar HC en éstos sujetos han sido bien caracterizados6,7,24- 26. La HC permite disminuir el porcentaje de grasa corporal e incrementar la masa magra; esto se asocia a menor riesgo cardiovascular. Después de pocas semanas de suspendido la HC se observa un menor gasto calórico y mayor incremento en la adiposidad27,28. El efecto de HC sobre la masa muscular es más notorio en hombres que en mujeres29,30.
Densidad mineral ósea
Los niños y adultos con deficiencia de HC tiene menor densidad mineral ósea que los sujetos sanos31-33. El tratamiento con HC en la fase de transición de los adolecentes con DHC puede permitir una mejor DMO. La acción anabólica de la HC sobre el hueso ha sido ampliamente estudiada y el impacto de HC en la masa ósea puede continuar incluso 1,5 años después de discontinuar la HC34.
Metabolismo de lípidos y carbohidratos
El impacto de HC en el perfil lipídico de niños y adolescentes es controvertido6,7. Algunos estudios no han mostrado beneficio en los parámetros lipídicos27,29,35. Si bien la mayoría de los reportes no muestran cambios en el metabolismo de los hidratos de carbono o en la sensibilidad insulínica, el tratamiento con HC puede, en algunos sujetos, producir insulino resistencia leve y transitoria13,26,27,30,35.
Fuerza muscular
En la mayoría de los estudios evaluados, la HC no ha mostrado aumentar la fuerza muscular1,36-38. La administración o la suspensión de HC en una cohorte de adolescentes con DHC en fase de transición no demostraron cambios significativos en la fuerza muscular11. Esto resultados son similares a los publicados en población adulta26,30 y adolescente27. La evidencia actual no apoyaría el uso de HC sólo con el objetivo de aumentar la fuerza muscular en este grupo etario.
Riesgo cardiovascular
Reportes de ensayos donde se utilizó HC en la fase de transición no mostraron un efecto sustancial en la función cardíaca30, pero la función endotelial y el grosor de la íntima mejoran en los adolescentes con DHC tratados versus los no tratados39. Los datos disponibles de población adulta, no han evidenciado un beneficio significativo en mejorar la función cardíaca o el rendimiento en ejercicio. Sin embargo, estudios recientes multicéntricos sugieren que la administración de HC durante 32 semanas a adultos deficitarios de esta hormona, no mejoró el rendimiento sub-máximo frente al ejercicio y no tuvo ningún impacto en la actividad física37.
Calidad de vida
Los resultados de los estudios de calidad de vida en personas con DHC han sido contradictorios, algunos han mostrado una mejoría en algunos parámetros de la calidad de vida, mientras que otros no27,40-44. Cuando se han utilizado cuestionarios específicamente diseñados para determinar calidad de vida en pacientes con DHC, el tratamiento con HC sí parece tener un impacto40; sin embargo, estos hallazgos parecen ser más importantes cuando la DHC se inicia en la vida adulta. Por otro lado, si consideramos que después de varios años de tratamiento con HC no hay diferencia de calidad de vida entre los sujetos sanos y con DHC, no se puede descartar que la HC haya tenido un rol en haber mantenido una calidad de vida normal.
Dosis de hormona de crecimiento
Frecuentemente es necesario utilizar dosis más altas durante la pubertad45, la dosis debe ser intermedia ente la dosis utilizada en la infancia y vida adulta. Este concepto se basa en el hecho que la producción de HC es alta en los adolescentes y va disminuyendo progresivamente en el adulto20.
Adolescentes y adultos jóvenes con DHC reciben un rango amplio de dosis durante la fase de transición, éstas fluctúan entre 12,5-20 ó incluso 25 ug/kg/día. La sugerencia de expertos es comenzar con una dosis de 12,5 ug/kg/día y luego continuar titulando con las concentraciones de IGF-1, manteniéndolas en el cuartil superior de lo normal para la edad17.
Seguimiento
Estos pacientes no sólo están en una transición hormonalmetabólica, también lo están desde el punto de vista emocional e intelectual. Es obligación del endocrinólogo pediátrico re-evaluar la función hipofisaria una vez alcanzada la talla final. Deben ser controlados periódicamente la antropometría (Peso, talla, perímetro de cintura), calidad de vida (encuesta de calidad de vida), IGF-1 y densitometría ósea (volumétrica de columna y cadera, especialmente en pacientes que tengan asociado hipogonadismo y usen glucocorticoides). Los intervalos entre controles quedan a discreción del médico tratante.
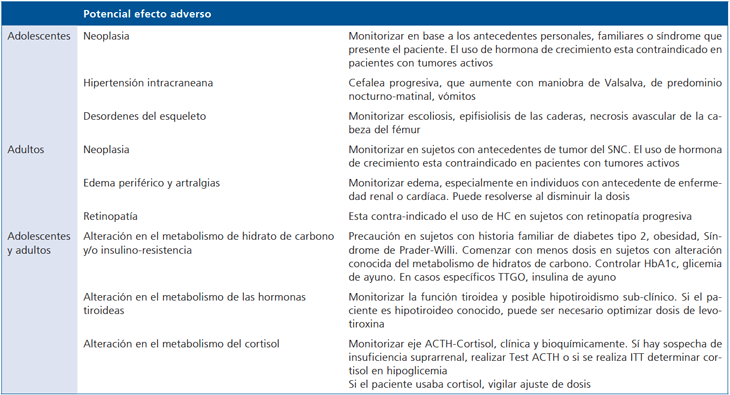
Potenciales efectos adversos
Durante los últimos 20 años han sido registrados más de 55.000 niños en el National Cooperative Growth Study (NCGS) en los cuales se han observado una incidencia de algún evento adverso tal como escoliosis, epifisiolisis de las caderas, hipertensión endocraneana, diabetes mellitus e insuficiencia suprarrenal en menos del 1%. En sujetos sin factores de riesgo no se ha observado un incremento de leucemias o de una nueva neoplasia o recurrencia de un tumor del sistema nervioso central46.
Se ha observado un aumento transitorio de la glicemia de ayuno, en adultos. Sin embargo, no existen evidencias que apoyen un aumento de riesgo de desarrollar diabetes tipo 2 en sujetos sin predisposición47-49.
Dado que el tratamiento con HC se ha asociado a una disminución de la T4-L, se sugiere monitorizar frecuentemente las hormonas tiroideas50.
La HC juega un rol importante en la modulación de los glucocorticoides a nivel periférico, principalmente inhibiendo la expresión de la enzima 11 beta-hidroxiesteroide dehidrogenasa 1 (11beta-HSD1) la cual actúa convirtiendo la cortisona en cortisol a nivel de los adipocitos e hígado, resultando en una menor generación local de cortisol. Por este motivo se sugiere tener un alto índice de sospecha de insuficiencia suprarrenal, ya que el tratamiento con HC puede desenmascarar una deficiencia compensada de glucocorticoides o hacer necesario aumentar la dosis de glucocorticoides en un individuo en tratamiento46. En la Tabla 2 se muestras otros eventos adversos relacionados con el uso de hormona de crecimiento.
Tabla 2. Monitorización de potenciales efectos adversos durante el tratamiento con hormona de crecimiento
Concluciones
- La mayoría de los sujetos con DHC tratados adecuadamente
durante la niñez, tienen una adecuada composición
corporal, densidad mineral ósea, función cardíaca, fuerza
muscular, metabolismo de carbohidratos y lípidos y de
calidad de vida.
- Los adolescentes con deficiencia aislada o idiopática de
HC requieren de re-evaluación cuidadosa de la secreción
de HC una vez completado el crecimiento lineal.
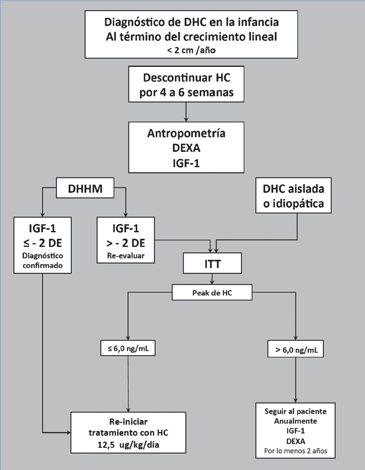
- Suspender tratamiento con HC durante 4 a 6 semanas
- Evaluar que estén con adecuada función tiroidea; y si corresponde, de glucocorticoides y esteroides sexuales, antropometría y edad ósea.
- Test de hipoglicemia (ITT) inducido con insulina, peak de GH menor a 6,0 ug/mL. d. Nivel basal de IGF-1 (no constituye criterio de diagnóstico, es importante para titular la dosis de HC).
- Densitometría ósea volumétrica.
- Los adolescentes con deficiencia de hormonas hipofisarias
múltiples o asociadas a alteraciones anatómicas de la
región hipotálamo hipofisaria no necesariamente requieren
re-evaluación de la secreción de HC.
- Suspender tratamiento con HC durante 4 a 6 semanas.
- Evaluar que estén con un adecuado remplazo hormonal en base a las deficiencias que existan, antropometría y edad ósea.
- Determinar concentración de IGF-1: i. Sí es menor o igual a -2 DE para la edad y sexo, se confirma diagnóstico de deficiencia de hormona de crecimiento. ii. Sí es mayor a -2 DE para la edad y sexo, solicitar (ITT)
- Test de hipoglicemia inducido con insulina, peak de GH menor o igual a 6 ng/mL
- Densitometría ósea volumétrica de columna y caderas basal y a los 18 meses del re-inicio del tratamiento.
- Dosis de hormona de crecimiento en fase de transición
- Re-iniciar HC a 12,5 ug/kg/día
- Ajustar dosis de HC en base a concentraciones de IGF- 1, mantener IGF-1 en cuartil superior de lo normal. La primera determinación de IGF-1 debe ser realizada a las 4 a 6 semanas hasta lograr la concentración objetivo, modificando la dosis de HC según respuesta. Luego de logrado la concentración de IGF-1 objetivo, continuar con determinaciones cada 6 meses.
-
Controlar TSH, T4-L, perfil lipídico, HbA1c, glicemia
cada 6 meses. d. Mantener dosis de transición hasta mediado de la segunda
década de vida.
Referencias
- Ho KK. 2007. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: a statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur J Endocrinol 157 (6): 695-700.
- 2. Shea HC, Levy RA. 2011. Transition Care of Growth Hormone
Deficient Patients from Pediatric Endocrinologists to Adult
Endocrinologists. Endocr Pract 8: 1-34.
- Geffner ME. 2009. Growth hormone replacement therapy:
transition from adolescence to adulthood. J Clin Res Pediatr
Endocrinol 1 (5): 205-208.
- Savage MO, Drake WM, Carroll PV, Monson JP. 2004.
Transitional care of GH deficiency: when to stop GH therapy.
Eur J Endocrinol 151 Suppl 1: S61-65.
- Matkovic V, Jelic T, Wardlaw GM, Ilich JZ, Goel PK, Wright JK,et al. 1994. Timing of peak bone mass in Caucasian females and its implication for the prevention of osteoporosis. Inference from a cross-sectional model. J Clin Invest 93 (2): 799-808.
- Molitch ME, Clemmons DR, Malozowski S, Merriam GR, Shalet SM, Vance ML, et al. 2006. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 91 (5): 1621-1634.
- Vance ML, Mauras N. 1999. Growth hormone therapy in adults and children. N Engl J Med 341 (16): 1206-1216.
- Norrelund H, Vahl N, Juul A, Moller N, Alberti KG, Skakkebaek NE, et al. 2000. Continuation of growth hormone (GH) therapy in GH-deficient patients during transition from childhood to adulthood: impact on insulin sensitivity and substrate metabolism. J Clin Endocrinol Metab 85 (5): 1912-1917.
- Drake WM, Carroll PV, Maher KT, Metcalfe KA, Camacho-Hubner C, Shaw NJ, et al. 2003. The effect of cessation of growth hormone (GH) therapy on bone mineral accretion in GH-deficient adolescents at the completion of linear growth. J Clin Endocrinol Metab 88 (4): 1658-1663.
- Shalet SM, Shavrikova E, Cromer M, Child CJ, Keller E, Zapletalova J, et al. 2003. Effect of growth hormone (GH) treatment on bone in postpubertal GH-deficient patients: a 2-year randomized, controlled, dose-ranging study. J Clin Endocrinol Metab 88 (9): 4124-4129.
- Mauras N, Pescovitz OH, Allada V, Messig M, Wajnrajch MP, Lippe B. 2005. Limited efficacy of growth hormone (GH) during transition of GH-deficient patients from adolescence to adulthood: a phase III multicenter, double-blind, randomized two-year trial. J Clin Endocrinol Metab 90 (7): 3946-3955.
- Tauber M, Moulin P, Pienkowski C, Jouret B, Rochiccioli P. 1997. Growth hormone (GH) retesting and auxological data in 131 GHdeficient patients after completion of treatment. J Clin Endocrinol Metab 82 (2): 352-356.
- Hartman ML, Crowe BJ, Biller BM, Ho KK, Clemmons DR, Chipman JJ. 2002. Which patients do not require a GH stimulation test for the diagnosis of adult GH deficiency? J Clin Endocrinol Metab 87 (2): 477-485.
- Maghnie M, Strigazzi C, Tinelli C, Autelli M, Cisternino M, Loche S, et al. 1999. Growth hormone (GH) deficiency (GHD) of childhood onset: reassessment of GH status and evaluation of the predictive criteria for permanent GHD in young adults. J Clin Endocrinol Metab 84 (4): 1324-1328.
- Gleeson HK, Gattamaneni HR, Smethurst L, Brennan BM, Shalet SM. 2004 Reassessment of growth hormone status is required at final height in children treated with growth hormone replacement after radiation therapy. J Clin Endocrinol Metab 89 (2): 662-666.
- Clayton PE, Cuneo RC, Juul A, Monson JP, Shalet SM, Tauber M. 2005. Consensus statement on the management of the GH-treated adolescent in the transition to adult care. Eur J Endocrinol 152 (2): 165-170.
- Radovick S, DiVall S. 2007. Approach to the growth hormonedeficient child during transition to adulthood. J Clin Endocrinol Metab 92 (4): 1195-1200.
- Zucchini S, Pirazzoli P, Baronio F, Gennari M, Bal MO, Balsamo A, et al. 2006. Effect on adult height of pubertal growth hormone retesting and withdrawal of therapy in patients with previously diagnosed growth hormone deficiency. J Clin Endocrinol Metab 91 (11): 4271-4276.
- Martha PM Jr, Rogol AD, Veldhuis JD, Kerrigan JR, Goodman DW, Blizzard RM. 1989. Alterations in the pulsatile properties of circulating growth hormone concentrations during puberty in boys. J Clin Endocrinol Metab 69 (3): 563-570.
- Veldhuis JD, Roemmich JN, Richmond EJ, Rogol AD, Lovejoy JC, Sheffield-Moore M, et al. 2005. Endocrine control of body composition in infancy, childhood, and puberty. Endocr Rev 26 (1): 114-146.
- Ho KK, Hoffman DM. 1995. Defining growth hormone deficiency in adults. Metabolism 44 (10 Suppl 4): 91-96.
- Secco A, di Iorgi N, Napoli F, Calandra E, Calcagno A, Ghezzi M, et al. 2009. Reassessment of the growth hormone status in young adults with childhood-onset growth hormone deficiency: reappraisal of insulin tolerance testing. J Clin Endocrinol Metab 94 (11): 4195-4204.
- Maghnie M, Aimaretti G, Bellone S, Bona G, Bellone J, Baldelli R, et al. 2005 Diagnosis of GH deficiency in the transition period: accuracy of insulin tolerance test and insulin-like growth factor-I measurement. Eur J Endocrinol 152 (4): 589-596.
- Bengtsson BA, Abs R, Bennmarker H, Monson JP, Feldt- Rasmussen U, Hernberg-Stahl E, et al. 1999. The effects of treatment and the individual responsiveness to growth hormone (GH) replacement therapy in 665 GH-deficient adults. KIMS Study Group and the KIMS International Board. J Clin Endocrinol Metab 84 (11): 3929-3935.
- Christiansen JS, Jorgensen JO. 1991. Beneficial effects of GH replacement therapy in adults. Acta Endocrinol (Copenh) 125 (1): 7-13.
- Mauras N, O’Brien KO, Welch S, Rini A, Helgeson K, Vieira NE, et al. 2000. Insulin-like growth factor I and growth hormone (GH) treatment in GH-deficient humans: differential effects on protein, glucose, lipid, and calcium metabolism. J Clin Endocrinol Metab 85 (4): 1686-1694.
- Vahl N, Juul A, Jorgensen JO, Orskov H, Skakkebaek NE, Christiansen JS. 2000. Continuation of growth hormone (GH) replacement in GH-deficient patients during transition from childhood to adulthood: a two-year placebo-controlled study. J Clin Endocrinol Metab 85 (5): 1874-1881.
- Cowan FJ, Evans WD, Gregory JW. 1999. Metabolic effects of discontinuing growth hormone treatment. Arch Dis Child 80 (6): 517-523.
- Attanasio AF, Howell S, Bates PC, Frewer P, Chipman J, Blum WF, et al. 2002. Body composition, IGF-I and IGFBP-3 concentrations as outcome measures in severely GH-deficient (GHD) patients after childhood GH treatment: a comparison with adult onset GHD patients. J Clin Endocrinol Metab 87 (7): 3368-3372.
- Underwood LE, Attie KM, Baptista J. 2003. Growth hormone (GH) dose-response in young adults with childhood-onset GH deficiency: a two-year, multicenter, multiple-dose, placebocontrolled study. J Clin Endocrinol Metab 88 (11): 5273-5280.
- Kaufman JM, Taelman P, Vermeulen A, Vandeweghe M. 1992.Bone mineral status in growth hormone-deficient males with isolated and multiple pituitary deficiencies of childhood onset. J Clin Endocrinol Metab 74 (1): 118-123.
- Holmes SJ, Economou G, Whitehouse RW, Adams JE, Shalet SM. 1994. Reduced bone mineral density in patients with adult onset growth hormone deficiency. J Clin Endocrinol Metab 78 (3): 669-674.
- Degerblad M, Bengtsson BA, Bramnert M, Johnell O, Manhem P, Rosen T, et al. 1995. Reduced bone mineral density in adults with growth hormone (GH) deficiency: increased bone turnover during 12 months of GH substitution therapy. Eur J Endocrinol 133 (2): 180-188.
- Biller BM, Sesmilo G, Baum HB, Hayden D, Schoenfeld D, Klibanski A. 2000. Withdrawal of long-term physiological growth hormone (GH) administration: differential effects on bone density and body composition in men with adult-onset GH deficiency. J Clin Endocrinol Metab 85 (3): 970-976.
- Johannsson G, Albertsson-Wikland K, Bengtsson BA. 1999. Discontinuation of growth hormone (GH) treatment: metabolic effects in GH-deficient and GH-sufficient adolescent patients compared with control subjects. Swedish Study Group for Growth Hormone Treatment in Children. J Clin Endocrinol Metab 84 (12): 4516-4524.
- Yarasheski KE, Zachwieja JJ, Campbell JA, Bier DM. 1995. Effect of growth hormone and resistance exercise on muscle growth and strength in older men. Am J Physiol 268 (2 Pt 1): E268-276.
- Hartman ML, Weltman A, Zagar A, Qualy RL, Hoffman AR, Merriam GR. 2008. Growth hormone replacement therapy in adults with growth hormone deficiency improves maximal oxygen consumption independently of dosing regimen or physical activity. J Clin Endocrinol Metab 93 (1): 125-130.
- Gotherstrom G, Bengtsson BA, Sunnerhagen KS, Johannsson G, Svensson J. 2005. The effects of five-year growth hormone replacement therapy on muscle strength in elderly hypopituitary patients. Clin Endocrinol (Oxf) 62 (1): 105-113.
- Szczepaniska Kostro J, Tolwinska J, Urban M, Gardziejczyk M, Glowinska B. 2004. Cardiac mass and function, carotid artery intima media thickness, homocysteine and lipoprotein levels in children and adolescents with growth hormone deficiency. J Pediatr Endocrinol Metab 17 (10): 1405-1413.
- Hull KL, Harvey S. 2003. Growth hormone therapy and Quality of Life: possibilities, pitfalls and mechanisms. J Endocrinol 179 (3): 311-333.
- Wiren L, Johannsson G, Bengtsson BA. 2001. A prospective investigation of quality of life and psychological well-being after the discontinuation of GH treatment in adolescent patients who had GH deficiency during childhood. J Clin Endocrinol Metab 86 (8): 3494-3498.
- Bjork S, Jonsson B, Westphal O, Levin JE. 1989. Quality of life of adults with growth hormone deficiency: a controlled study. Acta Paediatr Scand Suppl; 356: 55-59; discussion 60, 73-74.
- Rosen T, Wiren L, Wilhelmsen L, Wiklund I, Bengtsson BA. 1994. Decreased psychological well-being in adult patients with growth hormone deficiency. Clin Endocrinol (Oxf) 40 (1): 111-116.
- Abs R, Bengtsson BA, Hernberg-Stahl E, Monson JP, Tauber JP, Wilton P, et al. 1999. GH replacement in 1034 growth hormone deficient hypopituitary adults: demographic and clinical characteristics, dosing and safety. Clin Endocrinol (Oxf) 50 (6): 703-713.
- Mauras N, Attie KM, Reiter EO, Saenger P, Baptista J. High dose recombinant human growth hormone (GH) treatment of GH-deficient patients in puberty increases near-final height: a randomized, multicenter trial. Genentech, Inc., Cooperative Study Group. J Clin Endocrinol Metab 85 (10): 3653-3660.
- Bell J, Parker KL, Swinford RD, Hoffman AR, Maneatis T, Lippe B. 2010. Long-term safety of recombinant human growth hormone in children. J Clin Endocrinol Metab 95 (1): 167-177.
- Woodmansee WW, Hartman ML, Lamberts SW, Zagar AJ, Clemmons DR. 2010. Occurrence of impaired fasting glucose in GH-deficient adults receiving GH replacement compared with untreated subjects. Clin Endocrinol (Oxf) 72 (1): 59-69.
- Svensson J, Bengtsson BA. 2009. Safety aspects of GH replacement. Eur J Endocrinol 161 Suppl 1: S65-74.
- Attanasio AF, Jung H, Mo D, Chanson P, Bouillon R, Ho KK, et al. 2011. Prevalence and incidence of diabetes mellitus in adult patients on growth hormone replacement for growth hormone deficiency: a surveillance database analysis. J Clin Endocrinol Metab 96 (7): 2255-6122.
- Losa M, Scavini M, Gatti E, Rossini A, Madaschi S, Formenti I, et al. 2008. Long-term effects of growth hormone replacement therapy on thyroid function in adults with growth hormone deficiency. Thyroid 18 (12): 1249-1254.