Síndrome de Cushing secundario a secreción ectópica: un desafío diagnóstico
Jacques Young1 y María Consuelo Espinosa O.2
Cushing syndrome secondary to ectopic:
diagnostic challenge
1Servicio de Endocrinología y enfermedades de la reproducción, Hospital Bicêtre, París.
2Sección de Endocrinología Clínica Alemana. Sección de Endocrinología, Servicio de Medicina, Hospital Padre Hurtado.
Correspondencia:
M. Consuelo Espinosa O.
E-mail: espinosa.consue@gmail.com
Recibido: 18 de octubre de 2013
Aceptado: 21 de octubre de 2013
The diagnosis of Cushing Syndrome secondary to Ectopic ACTH secretion constitutes a challenge to the endocrinologist. The goal is to make a differential diagnosis of Cushing’s disease and localize the ACTH-secreting tumor, to achieve quick and effective management of a disease that can be fatal. The mainly diffuculties are the limited data due to their low prevalence and the wide variety of the origin tumors. Therefore, a comprehensive and multidisciplinary study is needed, analyzing each particular case. This article reviews the diagnostic alternatives, their strengths and weaknesses, proposing an algorithm that contributes to our clinical practice.
Key words: Cushing Syndrome, Neuroendocrine Tumors, Adrenocorticotropic Hormone.
El Síndrome de Cushing secundario a Secreción Ectópica (SCE) es una causa poco frecuente, pero potencialmente grave de hipercortisolismo ACTH dependiente. Su etiología más frecuente es un tumor neuroendocrino (TNE). Las series publicadas demuestran una prevalencia de 10 a 20% de los casos de Síndrome de Cushing (SC) ACTH dependientes1-9. Puede producir la muerte, tanto por los efectos hipercatabólicos, metabólicos e inmunológicos secundarios una secreción severa de cortisol, como por la enfermedad de base.
El SCE plantea 4 desafíos simultáneos al endocrinólogo:
1. Confirmación del diagnóstico.
2. Manejo rápido del hipercortisolismo a fin de mejorar estado general del paciente y prepararlo para el tratamiento definitivo de su enfermedad de base.
3. Tratamiento inmediato de las complicaciones asociadas.
4. Localización del tumor productor de ACTH, precisando su naturaleza y agresividad, para plantear el plan terapéutico multidisplinario.
El objetivo de esta breve revisión es aportar elementos de la literatura que permitan el mejor manejo de estos pacientes.
Etiología
Los tumores ectópicos secretores de ACTH son diversos desde el punto vista de su histología y localización, lo que dificulta su diagnóstico5,6,9,10,11 (Tabla 1). Sin embargo, cerca de la mitad de los casos se ubican en el tórax. En cuanto a la histología de estos tumores bronquiales existen 4 grandes tipos histológicos: Carcinoides típicos (TNE bien diferenciados), Carcinoides atípicos (TNE menos diferenciados) y TNE pobremente diferenciados de células grandes y pequeñas11-15.
La frecuencia relativa de cada uno de estos grupos es variable según la antigüedad del estudio y el modo de reclutamiento de los pacientes, dependiendo de si son series endocrinológicas u oncológicas. En las publicaciones endocrinológicas los tumores carcinoides son mucho más prevalentes que los carcinomas bronquiales de células pequeñas, estos últimos considerados como el prototipo de SCE paraneoplásico. En un estudio multicéntrico reciente sobre SCE en niños y jóvenes los TNE bien diferenciados son la causa más frecuente, al igual que en adultos16.
La pérdida de diferenciación se correlaciona con la agresividad tumoral, por lo que los carcinomas bronquiales de células grandes y pequeñas no constituyen un problema diagnóstico, sino presentan gran dificultad en la exeresis quirúrgica y el tratamiento coadyuvante. Por el contrario, los carcinoides bronquiales típicos pueden presentar un comportamiento cercano a la benignidad, son de pequeño tamaño (lo que puede dificultar su localización) y de crecimiento lento.
El cuadro clínico depende del grado de conservación de las características fenotípicas de la célula corticotropa17,18. Es decir, de la transcripción y maduración de la pro-opiomelanocortina (POMC) y de la presencia de receptores que regulan la secreción de ACTH (receptores de CRH, receptores V3 de la vasopresina y receptores de glucocorticoides). Por esta razón, los tumores carcinoides típicos pueden responder a la administración de análogos de la vasopresina, CRH o dexametasona in vivo e in vitro, tal como en la enfermedad de Cushing (EC)1,2,5,6,19. Esto explica las limitaciones de los test dinámicos al momento de distinguir entre EC y SCE.
En resumen, los TNE bien diferenciados son los que poseen el mayor problema diagnóstico, ya que pueden permanecer ocultos en las imágenes durante años y tener un patrón de secreción similar a lo observado en la EC.
Presentación clínica
Existen 2 formas de presentación clásicas: En el estudio de un SC ACTH dependiente o en un paciente oncológico con diagnóstico de neoplasia conocida, en el que aparece hipertensión arterial, hiperglicemia, hipokalemia, equimosis o empeoramiento del estado general.
A diferencia de la EC, que afecta principalmente a las mujeres, el SCE afecta de igual forma a hombres y mujeres1. La edad de presentación promedio es similar a la de la EC, aproximadamente 45 años11-20.
Clínicamente el SC es más intenso en el SCE, con mayor expresión de catabolismo (amiotrofia), edema de extremidades inferiores, hipokalemia con alcalosis metabólica, diabetes mellitus, hipertensión arterial y osteoporosis1-9. En el registro europeo de SC se encontró alta prevalencia de hirsutismo, diabetes mellitus y fracturas vertebrales con una frecuencia mayor a lo observado en EC (92% vs 63%, p < 0,05; 74% vs 33%, p < 0,01; 44% vs 25%, p < 0,05 respectivamente)20. La ganancia de peso está ausente en cerca de la mitad de los casos, existiendo pérdida ponderal en 10 a 20% de los casos11. La melanodermia es más frecuente que en la EC. En cerca del 50% de los casos hay infecciones oportunistas (cándida, herpes), las cuales empeoran el pronóstico7,21. Las trombosis venosas se documentan hasta en un 14% de los casos11. Como se ha mencionado, estos síntomas se asocian generalmente a tumores poco diferenciados, en cambio, la presentación clínica de un TNE bien diferenciado se asemeja más a la EC1-9,22.
La sobrevida está dada tanto por el tumor de origen como por las complicaciones del hipercortisolismo. En la serie oncológica del M.D. Anderson, que incluyó un 21% de carcinoma de células pequeñas, la mortalidad fue de 62,8%, siendo la progresión de la enfermedad y las infecciones oportunistas las principales causas de muerte (sobrevida promedio de 32 meses)11. En cambio, en la serie del National Institute of Health (NIH), que incluyó sólo un 3,3% de pacientes con carcinoma de células pequeñas, la mortalidad fue de 21%5.
Exámenes basales
El cortisol libre urinario (CLU) y la ACTH se encuentran en promedio más elevados en el SCE que en la EC. Sin embargo, existe una superposición de valores en ambas patologías, por lo que sólo son útiles valores muy elevados. La hipokalemia se observa hasta en un 72% de los casos11.
La anamnesis junto con el examen físico y los exámenes de laboratorio basales son de gran utilidad diagnóstica. La probabilidad de EC en una mujer de edad media que presenta un SC de instalación progresiva, con kalemia normal y CLU y ACTH moderadamente elevados, es cercana a 95%, con una especificidad entre 75 y 80%23. A la inversa, aproximadamente un 20% de los SCE se presentan de manera similar a la EC. De lo que se deduce que los estudios diagnósticos de segunda línea deberían tener una especificidad mayor al 90% para ser realmente útiles.
Se ha intentado medir la POMC, la beta endorfina y la relación LPH/ACTH plasmáticas (productos intermediarios de la secreción de ACTH), para relacionarla a la madura ción de la célula productora de ACTH y así distinguir entre EC y SCE. Sin embargo, existe superposición de valores24-26. Serían útiles en determinar la agresividad del tumor, tanto hipofisiario como ectópico.
Estudios dinámicos
Se sugiere utilizarlos como un complemento a la clínica y los exámenes de laboratorio basales para diferenciar entre EC y SCE. Poseen un valor diagnóstico débil, ya que se basan en estudios con un escaso número de pacientes portadores de SCE y los puntos de corte no son reproducibles en los diferentes laboratorios. Por otra parte, se debe tener en cuenta la gran variabilidad secretoria espontánea de cada paciente, por lo que es importante evaluar previamente el perfil de secreción espontánea de ACTH, cortisol plasmático y CLU del paciente, en varias muestras e idealmente hospitalizado. La realización de estos exámenes es engorrosa y lenta, por lo que no deben retardar el tratamiento del hipercortisolismo y sus complicaciones en el contexto de un paciente que puede agravarse rápidamente.
Test de frenación con dexametasona a dosis altas (Liddle)
En su forma clásica (8 mg/d en 2 días con una disminución de al menos del 50% del CLU) o en su forma rápida (8 mg a la medianoche con disminución de al menos del 50% del cortisol plasmático a las 8 AM), tiene una frecuencia de falsos positivos para EC que varía según el punto de corte entre 3% a más del 30%8,12,19,23. Asimismo, los falsos positivos en un carcinoide típico varían entre frecuencias muy bajas hasta la mitad de los pacientes1-9,22. Por otra parte, también existen falsos negativos, es decir, una EC que no frena1-9. Se han publicado numerosos estudios cuyo objetivo es determinar el punto de corte óptimo. En un estudio del NIH, la frenación del CLU de 90% determina una especificidad cercana al 100%, pero una sensibilidad de sólo 69%5. Otros estudios demuestran una utilidad mediocre, con una sensibilidad de 81% y una especificidad de 67%, siendo inferior a la combinación de clínica y exámenes basales23. Los grupos que aún recomiendan este test se basan en la importancia de realizarlo en un centro altamente especializado, con puntos de corte propios y considerando principalmente el cortisol plasmático más que el CLU5-6,27-28.
Test de estimulación con CRH
Consiste en la estimulación por CRH en forma iv y la medición de la respuesta de la ACTH y el cortisol. Es un test caro, no disponible en Chile. Un meta-análisis mostró una sensibilidad de 80% y una especificidad de 90% para el diagnóstico de EC, siendo mayor cuando la respuesta es exagerada. Muchos grupos continúan recomendándolo en asociación al test de frenación con dexametasona a dosis altas1-9,30,31.
Test de estimulación por desmopresina
El test consiste en administrar 10 μg de desmopresina (DDAVP) iv, midiendo la respuesta de ACTH a los 0,10, 20 y 30 min. El DDAVP es un agonista del receptor V2 de la vasopresina a nivel renal y presenta una afinidad débil por los receptores V1 (vasculares y hepáticos) y V3 (hipofisiarios). Contrariamente a lo observado en sujetos sanos, los tumores hipofisiarios secretores de ACTH expresan V2 y V3. Un aumento de ACTH de al menos 6 pmol/l orienta a EC. Sin embargo, un 30% de los tumores ectópicos expresan también receptores V3, por lo que su sensibilidad y especificidad son bajas (82% y 85-90% respectivamente)17,32,33.
Test de estimulación por metopirona
La metopirona es un inhibidor de la conversión de 11-deoxicortisol en cortisol, produciendo la consiguiente disminución del cortisol plasmático y del feedback negativo hipofisiario. Este test presenta las mismas debilidades que el test de dexametasona10.
Cateterismo de senos petrosos
Se basa en la gradiente de concentración de ACTH entre el sistema de drenaje de la hipófisis, es decir, de los senos petrosos (SP) y el sistema venoso periférico34. Debe ser realizado por un equipo experimentado35. La tolerancia del procedimiento es aceptable y puede ser realizado incluso en el contexto de un paciente grave y/o ante la urgencia de comenzar un tratamiento. Las complicaciones en centros especializados son menos del 0,5%35. Los niveles hormonales basales de ACTH demuestran una gradiente superior a 2 en la EC e inferior a 1,4 en el SCE. Sin embargo, según este criterio, 5 a 20% de los pacientes con EC no será diagnosticado34. La estimulación por CRH aumenta la sensibilidad, presentando una gradiente mayor o igual a 3 en EC y menor a 2 en SCE. En centros especializados, con un gran número de pacientes la especificidad es cercana al 95%, constituyendo el mejor examen para el diagnóstico etiológico del SC ACTH dependiente1-9,36. Las interpretaciones erradas son generalmente falsos negativos, es decir, baja gradiente en casos portadores de EC, debido a variantes anatómicas del sistema venoso y a la ubicación incorrecta de los catéteres34-36. Por lo tanto, la ausencia de gradiente no diagnostica necesariamente un SCE y si no existe ninguna lesión ectópica identificada, el seguimiento de imágenes deberá realizarse también a nivel hipofisiario. Por el contrario, los falsos positivos son raros. Estos pueden explicarse por una secreción de cortisol intermitente, por secreción de CRH del TNE o por tratamiento con fármacos anticortisólicos, por lo que es indispensable que el paciente se encuentre en hipercortisolismo al momento del examen37. En los países en que la CRH no está disponible, como en Chile, la DDAVP ha demostrado resultados comparables38,39.
Imágenes
La localización más frecuente de los TNE productores de SCE es toráxica (más del 50% de los casos), por lo que un scanner de tórax es la primera imagen a realizar. Debido al pequeño tamaño y a la localización próxima a los vasos pulmonares de los carcinoides bronquiales, el radiólogo debe buscar dirigidamente esa zona, mediante un TAC helicoidal con cortes finos (menor a 1,2 mm)40. Los carcinomas medulares de tiroides son fácilmente identificados por la ecografía. Si el estudio es negativo, se debe continuar con TAC o RM abomino-pelviana, siendo los TNE de esta localización generalmente de mayor tamaño y por lo tanto, fácilmente identificables en las imágenes convencionales. En casos graves, se realiza una imagen de tórax, abdomen y pelvis en primera línea. En esta primera etapa se localizan 2/3 de los casos. La ecografía endoscópica pancreática es discutible, ya que los tumores pancreáticos secretores de ACTH rara vez permanecen ocultos1.
En una segunda etapa se podría indicar un octreoscan (In 111- pentreotide), cintigrama que capta los receptores de somatostatina tipo 2. Este examen puede pesquisar carcinoides bronquiales hasta de 6 mm. El beneficio como examen aislado del octreoscan es inferior al de las imágenes convencionales, por lo que se debe tener precaución al diagnosticar imágenes cintigráficas que no tienen representación en TAC o RM5,6,41,42. Muchos autores no recomiendan su indicación en pacientes con TAC o RM negativos, ya que no aportaría mayor información. En la serie del MD Anderson el octreoscan fue negativo en todos los pacientes con tumor oculto a las imágenes convencionales11.
Al complementar imágenes anatómicas (RM o TAC) con imágenes funcionales se mejora el valor predictivo positivo (VPP) para localizar el SCE. En una revisión de 30 pacientes con SCE, el VPP de la RM y TAC aumentó al combinarlas con el octreoscan (de 66 y 74% a 91 y 100% respectivamente)40. El F-DOPA-PET tiene un alto VPP al combinarlo con imágenes anatómicas (100%), sin embargo, tiene baja sensibilidad (55%) y no está disponible en nuestro medio40.
El PET-FDG es útil en tumores menos diferenciados y más agresivos43. Está indicado en la etapificación de cáncer bronquial o en feocromocitoma. En tumores ocultos no mejora su localización, ya que estos metabólicamente son menos activos11,40.
A pesar de un estudio de localización extenso hasta cerca de un 20% de los SCE permanecen ocultos, siendo generalmente carcinoides bronquiales que pueden evidenciarse en meses o incluso en más de 10 años1-9,11. En la serie del NIH el diámetro promedio de los carcinoides bronquiales era de 11 mm y la localización luego del inicio del SC en promedio fue a los 5,2 años5. Actualmente, los tumores realmente ocultos son cada vez menos, debido a la mejoría de las imágenes y la especialización de los centros de referencia.
En estos casos, se sugiere tratar el hipercortisolismo y controlar con imágenes cada 6 meses. La repetición del octreoscan es discutible42. Cabe destacar, que existe la hiperplasia tímica secundaria a un tratamiento eficaz del hipercortisolismo que puede simular un TNE del timo44.
Por otra parte, la RM hipofisiaria es un examen accesible y ampliamente utilizado para descartar un adenoma corticotropo, sin embargo, tiene las desventajas de que los adenomas son muchas veces difíciles de visualizar (tamaño promedio 4-5 mm) y de la alta prevalencia de adenomas no funcionantes menores de 5 mm45. La sensibilidad en las distintas series va de 50 a 70% y los falsos positivos, de 0 a 20%5,6,23. Por lo tanto, una RM positiva no afirma el diagnostico de EC, principalmente cuando el tamaño es menor a 5 mm.
Proceso diagnóstico
Actualmente, no existe un algoritmo diagnóstico único para determinar la etiología del SC ACTH dependiente. Proponemos un estudio práctico, analizando en conjunto todos los elementos diagnósticos (clínica, laboratorio e imágenes) y considerando tanto su disponibilidad en nuestro medio, como sus fortalezas y debilidades.
Se deben distinguir 2 situaciones clínicas diferentes:
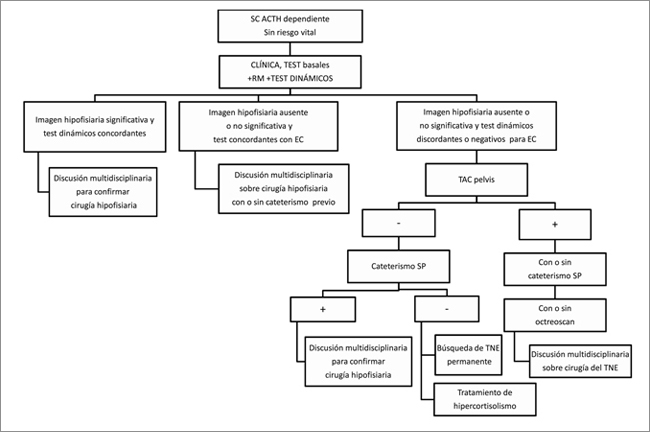
La primera, en la cual el paciente no presenta riesgo vital y puede ser estudiado en forma exhaustiva, sin apremio (Figura 1). En este paciente con diagnóstico apropiado de SC ACTH dependiente (clínica compatible, hipercortisolismo y ACTH no disminuida), es conveniente repetir los exámenes hormonales basales, tanto para asegurar el diagnóstico como para poder interpretar de mejor manera los tests dinámicos. En cuanto a estos estudios, se sugiere realizar los que estén disponibles según el centro y la experiencia clínica, teniendo en cuenta sus limitaciones particulares. Los tests de estimulación deberían ser realizados en primer lugar (CRH-DDAVP) y posteriormente los test de frenación para disminuir errores de interpretación. La RM hipofisiaria puede ser realizada al mismo tiempo. Si se demuestra una imagen hipofisiaria significativa, es decir, compatible con adenoma y de un tamaño que arbitrariamente se ha estimado en mayor o igual a 5 ó 6 mm, acompañada de tests dinámicos concordantes, la cirugía debe ser discutida por un equipo multidisciplinario (endocrinólogos, neurocirujanos, neuroradiólogos, etc.). En el caso de que no exista imagen significativa y sin embargo, la clínica, los exámenes de laboratorio y los test dinámicos orientan a una EC, se discutirá en un equipo multidisciplinario la posibilidad de realizar una exploración quirúrgica hipofisiaria. Esto consiste en realizar una adenomectomía, cuando el cirujano identifica el tumor, o una hipofisectomía (2/3 posteriores, lugar donde habitualmente se encuentran adenomas productores de ACTH) cuando no lo observa. Esta conducta en manos de expertos mejora el pronóstico y no produce más complicaciones que la cirugía de un tumor identificado previamente por RM46,47. Si en la RM no se observa imagen o ésta no es significativa y los test dinámicos son discordantes o no compatibles con EC, se continúa el estudio con scanner de tórax con cortes finos y luego de abdomen y pelvis (TAC T-A-P). En caso de observar una imagen sugerente, se discutirá en equipo multidisciplinario la resección del TNE. La utilidad del octreoscan a este nivel es moderada, por lo que deberá ser realizado según su disponibilidad. En caso de no encontrar imagen, se sugiere realizar un cateterismo de SP (algunos grupos lo realizan en todos los pacientes, aunque se encuentre imagen sugerente). El octreoscan en este caso aporta poca información. Si el cateterismo es concordante con EC, se discutirá la cirugía hipofisaria en equipo multidisciplinario. Si el cateterismo es negativo y fue técnicamente bien realizado, se debe efectuar una búsqueda de tumor ectópico sistemática a largo plazo y además, tratar el hipercortisolismo en sus diferentes modalidades. Es importante destacar la importancia del tratamiento médico, ya que si es bien tolerado puede constituir una herramienta muy útil en el manejo sintomático y profiláctico de complicaciones.
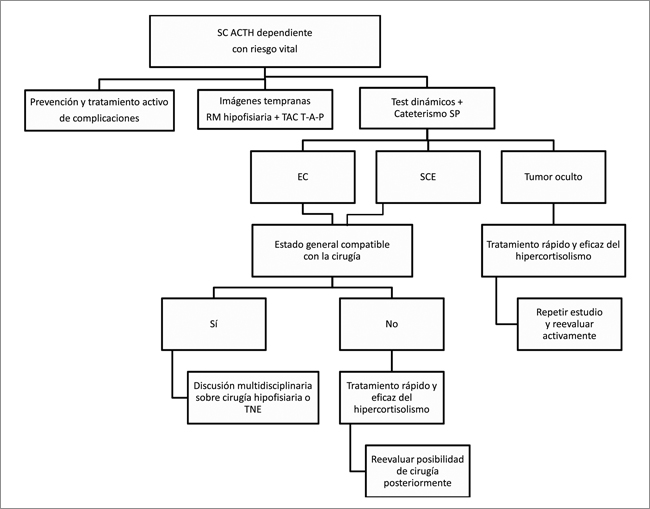
La segunda situación clínica se refiere al paciente con riesgo vital, en el cual es importante que el estudio sea rápido y efectivo, y se realice en concomitancia con la prevención y el tratamiento activo de las complicaciones del hipercortisolismo (Figura 2). Los test dinámicos y las imágenes pueden ser realizados al mismo tiempo. El cateterismo de SP en este caso generalmente no presenta mayor riesgo que el habitual, siempre que la prevención y el tratamiento de las complicaciones sea adecuado. Posteriormente, con el diagnóstico de EC o SCE, se discutirá la posibilidad de la cirugía dependiendo de la gravedad del paciente. Si ésta no es posible, se debe tratar medicamente el SC, escogiendo idealmente el fármaco con el efecto más rápido y eficaz posible. Una vez que el paciente esté estabilizado, se debe re-evaluar la posibilidad de la cirugía.
En caso de que el tumor permanezca oculto, se debe igualmente tratar el hipercortisolismo, revisar el estudio ya efectuado, repetirlo en caso necesario y mantener controles clínicos e imagenológicos seriados.
Conclusión
El diagnóstico de SCE es complejo, ya que es un síndrome poco frecuente que puede estar ligado a gran variedad de tumores y sus síntomas se pueden confundir con aquellos del tumor de origen. Además, las series publicadas son escasas, por lo que no existen puntos de corte específicos a nivel de los exámenes de laboratorio que lo diferencien claramente de una EC. Su localización puede ser difícil por tratarse generalmente de tumores pequeños, requiriendo un estudio imagenológico exhaustivo. Por otra parte, su morbi-mortalidad es alta en algunos casos, por lo que se requiere un manejo rápido y eficaz a fin de evitar complicaciones agudas. Creemos que el algoritmo diagnóstico propuesto constituye una guía que puede aplicarse a nuestra realidad local.
En resumen, el proceso diagnóstico del SCE es un desafío para el endocrinólogo y debe ser exhaustivo, apoyado en un equipo multidisciplinario y basado en las posibilidades y experiencia de cada centro, analizando cada caso en forma particular.
Referencias bibliográficas
- Beuschlein F, Hammer GD. 2002. Ectopic pro-opiomelanocortin syndrome. Endocrinol Metab Clin North Am 31: 191-234.
- Wajchenberg BL, Mendonca BB, Liberman B, Pereira MA, Carneiro PC, Wakamatsu A, et al. 1994. Ectopic adrenocorticotropic hormone syndrome. Endocr Rev 15: 752-787.
- Becker M, Aron DC. 1994. Ectopic ACTH syndrome and CRH-mediated Cushing’s syndrome. Endocrinol Metab Clin North Am 23: 585-606.
- Newell-Price J, Trainer P, Besser M, Grossman A. 1998. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev 19: 647-672.
- Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. 2005. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab 90: 4955-4962.
- Isidori AM, Kaltsas GA, Pozza C, Frajese V, Newell-Price J, Reznek RH, et al. 2006. The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. J Clin Endocrinol Metab 91: 371-377.
- Invitti C, Pecori Giraldi F, de Martin M, Cavagnini F. 1999. Diagnosis and management of Cushing’s syndrome: results of an Italian multicentre study. Study Group of the Italian Society of Endocrinology on the Pathophysiology of the Hypothalamic-Pituitary-Adrenal Axis. J Clin Endocrinol Metab 84: 440-448.
- Boscaro M, Arnaldi G. 2009. Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab 94: 3121-3131.
- Aniszewski JP, Young WF Jr, Thompson GB, Grant CS, van Heerden JA. 2001. Cushing syndrome due to ectopic adrenocorticotropic hormone secretion. World J Surg 25: 934-940.
- Tabarin A. 2007. Sécrétion ectopique d’ACTH. Diagnostic et thérapeutique. En: Chanson P, Young J. Traité d’endocrinologie. Paris, Editorial Flammarion 339.
- Ejaz S, Vassilopoulou-Sellin R, Busaidy NL, Hu MI, Waguespack SG, Jiménez C, et al. 2011. Cushing syndrome secondary to ectopic adrenocorticotropic hormone secretion: the University of Texas MD Anderson Cancer Center Experience. Cancer 117: 4381-4389.
- Travis WD, Linnoila RI, Tsokos MG, Hitchcock CL, Cutler GB Jr, Nieman L. 1991. Neuroendocrine tumors of the lung with proposed criteria for large-cell neuroendocrine carcinoma. An ultrastructural, immunohistochemical, and flow cytometric study of 35 cases. Am J Surg Pathol 15: 529-553.
- Hage R, de la Rivière AB, Seldenrijk CA, van den Bosch JM. 2003. Update in pulmonary carcinoid tumors: a review article. Ann Surg Oncol 10: 697-704.
- Kaltsas GA, Besser GM, Grossman AB. 2004. The diagnosis and medical management of advanced neuroendocrine tumors. Endocr Relat Cancer 11: 523-535.
- Baudin et al. 2008. Tumeurs neuroendocrines thoraxiques et digestives. En: Baudin, Ducreux. Tumeurs neuroendocrines thoraxiques et digestives. Paris, Editorial Springer-Verlag.
- More J, Young J, Reznik Y, Raverot G, Borson-Chazot F, Rohmer V, et al. 2011. Ectopic ACTH syndrome in children and adolescents. J Clin Endocrinol Metab 96: 1213-1222.
- De Keyser Y, et al. 2001. Adrenal disorders. En: Margioris N, Chrousos G. Adrenal disorders. Totowa, New Jersey. Editorial human press 165.
- Messager M, Carrière C, Bertagna X, de Keyzer Y. 2006. RT-PCR analysis of corticotroph-associated genes expression in carcinoid tumours in the ectopic-ACTH syndrome. Eur J Endocrinol 154: 159-166.
- Florkowski CM, Wittert GA, Lewis JG, Donald RA, Espiner EA. 1994. Glucocorticoid responsive ACTH secreting bronchial carcinoid tumours contain high concentrations of glucocorticoid receptors. Clin Endocrinol (Oxf) 40: 269-274.
- Valassi E, Santos A, Yaneva M, Tóth M, Strasburger CJ, Chanson P, et al. 2011. The European Registry on Cushing’s syndrome: 2-year experience. Baseline demographic and clinical characteristics. Eur J Endocrinol 165: 383-392.
- Sarlis NJ, Chanock SJ, Nieman LK. 2000. Cortisolemic indices predict severe infections in Cushing syndrome due to ectopic production of adrenocorticotropin. J Clin Endocrinol Metab 85: 42-47.
- Limper AH, Carpenter PC, Scheithauer B, Staats BA. 1992. The Cushing syndrome induced by bronchial carcinoid tumors. Ann Intern Med 117: 209-214.
- Aron DC, Raff H, Findling JW. 1997. Effectiveness versus efficacy: the limited value in clinical practice of high dose dexamethasone suppression testing in the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab 82: 1780-1785.
- Tabarin A, Corcuff JB, Rashedi M, Angibeau R, Caille JM, Ducassou D, et al. 1992. Multihormonal response to corticotropin-releasing hormone in inferior petrosal sinus blood of one patient with Cushing’s disease: comparison with in vitro secretion of the tumoral corticotropes. Acta Endocrinol (Copenh) 127: 284-288.
- Raffin-Sanson ML, Massias JF, Dumont C, Raux-Demay MC, Proeschel MF, Luton JP, et al. 1996. High plasma proopiomelanocortin in aggressive adrenocorticotropin-secreting tumors. J Clin Endocrinol Metab 81: 4272-4277.
- Kuhn JM, Proeschel MF, Seurin DJ, Bertagna XY, Luton JP, Girard FL. 1989. Comparative assessment of ACTH and lipotropin plasma levels in the diagnosis and follow-up of patients with Cushing’s syndrome: a study of 210 cases. Am J Med 86: 678-684.
- Flack MR, Oldfield EH, Cutler GB Jr, Zweig MH, Malley JD, Chrousos GP, et al. 1992. Urine free cortisol in the high-dose dexamethasone suppression test for the differential diagnosis of the Cushing syndrome. Ann Intern Med 116: 211-217.
- Isidori AM, Kaltsas GA, Mohammed S, Morris DG, Jenkins P, Chew SL, et al. 2003. Discriminatory value of the low-dose dexamethasone suppression test in establishing the diagnosis and differential diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 88: 5299-5306.
- Newell-Price J, Morris DG, Drake WM, Korbonits M, Monson JP, Besser GM, Grossman AB. 2002. Optimal response criteria for the human CRH test in the differential diagnosis of ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab 87: 1640-1645.
- Nieman LK, Oldfield EH, Wesley R, Chrousos GP, Loriaux DL, Cutler GB Jr. 1993. A simplified morning ovine corticotropin-releasing hormone stimulation test for the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab 77: 1308-1312.
- Malchoff CD, Orth DN, Abboud C, Carney JA, Pairolero PC, Carey RM. 1988. Ectopic ACTH syndrome caused by a bronchial carcinoid tumor responsive to dexamethasone, metyrapone, and corticotropin-releasing factor. Am J Med 84: 760-764.
- Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. 2008. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 93: 1526-1540.
- Terzolo M, Reimondo G, Alì A, Borretta G, Cesario F, Pia A, et al. 2001. The limited value of the desmopressin test in the diagnostic approach to Cushing’s syndrome. Clin Endocrinol (Oxf) 54: 609-616.
- Tabarin A, Greselle JF, San-Galli F, Leprat F, Caille JM, Latapie JL, et al. 1991. Usefulness of the corticotropin-releasing hormone test during bilateral inferior petrosal sinus sampling for the diagnosis of Cushing’s disease. J Clin Endocrinol Metab 73: 53-59.
- Kaltsas GA, Giannulis MG, Newell-Price JD, Dacie JE, Thakkar C, Afshar F, et al. 1999. A critical analysis of the value of simultaneous inferior petrosal sinus sampling in Cushing’s disease and the occult ectopic adrenocorticotropin syndrome. J Clin Endocrinol Metab 84: 487-492.
- Lindsay JR, Nieman LK. 2005. Differential diagnosis and imaging in Cushing’s syndrome. Endocrinol Metab Clin North Am 34: 403-421.
- Young J, Deneux C, Grino M, Oliver C, Chanson P, Schaison G. 1998. Pitfall of petrosal sinus sampling in a Cushing’s syndrome secondary to ectopic adrenocorticotropin-corticotropin releasing hormone (ACTH-CRH) secretion. J Clin Endocrinol Metab 83: 305-308.
- Deipolyi AR, Hirsch JA, Oklu R. 2013. Bilateral inferior petrosal sinus sampling with desmopressin. J Neurointerv Surg 5: 487-488.
- Castinetti F, Morange I, Dufour H, Jaquet P, Conte-Devolx B, Girard N, et al. 2007. Desmopressin test during petrosal sinus sampling: a valuable tool to discriminate pituitary or ectopic ACTH-dependent Cushing’s syndrome. Eur J Endocrinol 157: 271-277.
- Zemskova MS, Gundabolu B, Sinaii N, Chen CC, Carrasquillo JA, Whatley M, et al. 2010. Utility of various functional and anatomic imaging modalities for detection of ectopic adrenocorticotropin-secreting tumors. J Clin Endocrinol Metab 95: 1207-1219.
- Torpy DJ, Chen CC, Mullen N, Doppman JL, Carrasquillo JA, Chrousos GP, Nieman LK. 1999. Lack of utility of (111). In-pentetreotide scintigraphy in localizing ectopic ACTH producing tumors: follow-up of 18 patients. J Clin Endocrinol Metab 84: 1186-1192.
- Tabarin A, Valli N, Chanson P, Bachelot Y, Rohmer V, Bex-Bachellerie V, et al. 1999. Usefulness of somatostatin receptor scintigraphy in patients with occult ectopic adrenocorticotropin syndrome. J Clin Endocrinol Metab 84: 1193-1202.
- Pacak K, Ilias I, Chen CC, Carrasquillo JA, Whatley M, Nieman LK. 2004. The role of [(18)F] fluorodeoxyglucose positron emission tomography and [(111)In]-diethylenetriaminepentaacetate-D-Phe-pentetreotide scintigraphy in the localization of ectopic adrenocorticotropin-secreting tumors causing Cushing’s syndrome. J Clin Endocrinol Metab 89: 2214-2221.
- Tabarin A, Catargi B, Chanson P, Hieronimus S, Corcuff JB, Laurent F, et al. 1995. Pseudo-tumours of the thymus after correction of hypercortisolism in patients with ectopic ACTH syndrome: a report of five cases. Clin Endocrinol (Oxf) 42: 207-213.
- Hall WA, Luciano MG, Doppman JL, Patronas NJ, Oldfield EH. 1994. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med 120: 817-820.
- Salenave S, Gatta B, Pecheur S, San-Galli F, Visot A, Lasjaunias P, et al. 2004. Pituitary magnetic resonance imaging findings do not influence surgical outcome in adrenocorticotropin-secreting microadenomas. J Clin Endocrinol Metab 89: 3371-3376.
- Sheth SA, Mian MK, Neal J, Tritos NA, Nachtigall L, Klibanski A, Biller BM, Swearingen B. 2012. Transsphenoidal surgery for cushing disease after nondiagnostic inferior petrosal sinus sampling. Neurosurgery 71: 14-22.