Carcinoma adrenal virilizante en una adolescente. Reporte de caso
Gabriela Mintegui1*, Nasthia Quilismal2, Beatriz Mendoza3, Patricia Agüero4.
Virilizing adrenal carcinoma in an adolescent. Case report
- Médico endocrinólogo, Profesora agregada, Clínica de Endocrinología y Metabolismo del Hospital de Clínicas. Facultad de Medicina. U de la R. Montevideo, Uruguay.
- Médico. Postgrado endocrinología, Clínica de endocrinología y metabolismo del Hospital de Clínicas. Facultad de Medicina. U de laR. Montevideo, Uruguay.
- Médico endocrinólogo, Profesora. Directora, Clínica de endocrinología y metabolismo del Hospital de Clínicas. Facultad de Medicina. U de la R. Montevideo, Uruguay.
- Médico endocrinólogo, Asistente, Clínica
de endocrinología y metabolismo del
Hospital de Clínicas. Facultad de Medicina.
U de la R. Montevideo, Uruguay.
*Correspondencia: Gabriela Mintegui / gabymin92@gmail.com Dirección: Emilio Frugoni 1199, apto 601. CP. 11200 Montevideo, Uruguay.
No se recibió ayuda financiera.
Recibido: 08-08-2022.
Aceptado: 12-09-2022.
Resumen: Los tumores suprarrenales virilizante son infrecuentes y representan
5-6% de los tumores de esas glándulas1. Pueden secretar diferentes andrógenos
como dehidroepiandrosterona sulfato (DHEAS), androstenediona y testosterona.
Las características clínicas dependen de la edad de presentación; en niños pueden
determinar pubertad precoz y en mujeres en edad fértil ocasionar hirsutismo, amenorrea
o ciclos oligomenorreicos y diversos grados de virilización2. Los carcinomas
adrenocorticales son tumores raros y la incidencia es aproximadamente uno a dos
por millón de habitantes/año3,4. Los exámenes de imagen como la tomografía o la
resonancia confirman el origen suprarrenal, valoran la presencia de metástasis y
definen la conducta terapéutica5. La presentación inicial en pacientes pediátricos
mayoritariamente es con virilización6 y aproximadamente el 50% de los pacientes
adultos con carcinoma adrenal tienen un estadio de la enfermedad relativamente
avanzado7. El tratamiento de elección es la cirugía y sigue siendo la mejor esperanza
para la supervivencia a largo plazo8. El pronóstico habitual para el carcinoma
adrenocortical es generalmente malo con una supervivencia global a 5 años de 20
a 25%5 en adultos, pero en niños y adolescentes la supervivencia puede llegar al
100%9. Se presenta el caso de una paciente con fenotipo totalmente masculino
con diagnóstico de carcinoma adrenal virilizante que comienza en la infancia y se
diagnostica en la adolescencia.
Palabras clave: Carcinoma adrenal; Clitoromegalia; Suprarrenales; Virilización.
Abstract: Virilizing adrenal tumors are uncommon and represent 5-6% on tumors of
these glands1. They can secrete different androgens such as dehydroepiandrosterone
sulfate (DHEAS), androstenedione, and testosterone. Clinical features depend on
the age of presentation; in children they can determine precocious puberty and in
women of childbearing age cause hirsutism, amenorrhea or oligomenorrheic cycles
and various degrees of virilization2. Diagnosis consists of clinical evidence of hyperandrogenism,
accompanied by an increase in androgens in the blood, especially
DHEAS, whose origin is mainly adrenal. Adrenocortical carcinomas are rare and the
incidence is approximately one to two per million inhabitants/year3,4. Imaging tests
such as tomography or resonance confirm the adrenal origin, assess the presence
of metastases and define the therapeutic approach5. In initial presentation in most
pediatric patients is with virilization6 and approximately 50% adult’s patients with
adrenal carcinoma have a relatively advanced stage of the disease7. The treatment
of choice is surgery and is the best hope for long-term survival. The usual prognosis for adrenocortical carcinoma is generally poor with a 5-year overall survival of 20 to
25%5 in adults, but in children and adolescent’s survival can reach 100%9. We present
the case of a patient with a totally male phenotype diagnosed with virilizing adrenal
carcinoma that begins in childhood and is diagnosed in adolescence.
Keywords: Adrenal carcinoma; Adrenals virilization; Clitoromegaly.
Introducción
La mayoría de tumores adrenocorticales son adenomas benignos no funcionantes. Los adenomas benignos secretores de hormonas causan síndrome de Cushing, aldosteronismo primario o con mucha menor frecuencia, virilización3,5. Los carcinomas adrenocorticales (CAC) son tumores raros, generalmente esporádicos y a menudo agresivos; pueden ser funcionantes y causar virilización2. Aparecen a cualquier edad, pero se plantea una distribución etaria bimodal, con picos en la infancia antes de los cinco años y en la cuarta o quinta década de vida3. Generalmente el grado de agresividad y el ritmo de progresión de la enfermedad son más rápidos en adultos que en niños5. Las mujeres desarrollan CAC con más frecuencia que los hombres (relación mujer-hombre de 1,5 a 2,5:1)10. Los pacientes pediátricos con carcinomas adrenales asocian síndromes genéticos como el síndrome de Beckwith- Wiedemann11. El objetivo es mostrar el caso de un tumor con características anatomopatológicas de malignidad, pero con un comportamiento clínico indolente en cuanto al compromiso de otras estructuras.
Presentación de caso
Adolescente de 16 años con antecedentes de pubarquia a los 3 años de edad, permanente y progresiva, con 6 años vello axilar, luego acné, seborrea y a los 11 vello facial. Luego clitoromegalia; negó cambios en la voz u otros elementos de hiperandrogenismo clínico. Consultó por amenorrea primaria. Al examen: peso 57 kg, talla 162 cm, hábito masculino. Acné severo en rostro y dorso, seborrea. Vello terminal en mentón, labio superior, axilas, línea infraumbilical, región sacra y muslos (Figuras 1A y 1B); escala de Ferriman-Galleway: 18. Mamas Tanner 1, clitoromegalia, vello púbico Tanner V (Figura 1C). Se palpó tumoración a nivel de hipocondrio y flanco derecho de 15 cm, dura, indolora. Laboratorio: testosterona total: 1478 ng/dl (6-82), testosterona libre: 26,08 mcg/mL (0,10- 3,20), DHEAS: 3590 mcg/dl (0-140), 17OH Progesterona 12,4 ng/ml (0,03-0,91). Cortisol basal, cortisol post 1 mg de dexametasona, ionograma normales. Ecografía ginecológica: útero adulto normal, endometrio 2 mm, ovarios: múltiples quistes. Tomografía (Figura 2A) que mostró voluminosa masa redondeada de bordes bien definidos, de 16 x 10 x 9,5 cm. Densidad sólida, heterogénea, con sectores hipodensos y calcificaciones; desciende y rota riñón derecho. Comprime y desplaza vena cava inferior. Hígado desplazado hacia adelante, sin alteración de la densidad hepática. No adenomegalias abdomino-pélvicas. RNM (Figura 2B): tumoración sólida suprarrenal derecha de 17 x 10 x 10 cm, con múltiples focos de grasa y algunas áreas sugestivas de necrosis o quísticas. Presentó realce inhomogéneo; contornos bien delimitados. Se realizó adrenalectomía derecha sin complicaciones. Patología, macroscopía: pieza de resección de 746 gramos, superficie externa lisa, completamente encapsulada (Figura 2C). Al corte, extensas áreas de necrosis y hemorragias; extensas áreas de calcificación. Microscopía: proliferación celular atípica pleomórfica que crece en patrón sólido trabecular, constituida por células grandes poliédricas, abundante citoplasma eosinófilo finamente granular (15% de células tumorales con citoplasma claro), núcleos pleomórficos con nucléolos con nucléolos evidentes e inclusiones intranucleares. Figuras abigarradas, mitosis típicas y atípicas. Múltiples focos de tejido hematopoyético. Se observan embolias a nivel vascular. En cuanto al índice mitótico, se observaron 30 mitosis por campos de gran aumento (CGA) con un Ki 67 del 12%. En suma: neoplasia primitiva suprarrenal maligna, con los caracteres de un carcinoma de corteza suprarrenal, criterios de Weiss 6 puntos. Al mes presentó disminución de clitoromegalia, a los 3 meses retomó ciclos menstruales mensuales y disminución del vello luego de depilación. A los 2 años de la cirugía se embarazó y tuvo un parto sin complicaciones. Desconocemos evolución posterior porque la paciente no volvió a consulta.
Discusión
En la presentación clínica, aproximadamente el 60% de los CAC producen síndrome de hiperandrogenismo12. Los adultos con CAC funcionantes presentan síndrome de Cushing en 45%, cosecreción mixta de Cushing y virilización en 25%3,12 y menos del 10% presentan virilización exclusivamente. Los niños suelen presentar virilización con mayor frecuencia, en el 84% de los casos6,13.
En mujeres el exceso de andrógenos ocasiona: hirsutismo, diversos grados de virilización (clitoromegalia, atrofia mamaria, voz más grave, alopecia androgénica, aumento del tejido muscular) y trastornos menstruales (amenorrea primaria o secundaria)2. En este caso generó un cambio total y un fenotipo totalmente masculino. El exceso de secreción de DHEA y DHEAS define el hiperandrogenismo suprarrenal; el hirsutismo y la virilización son causados por los andrógenos más potentes como la androstenediona y la testosterona.
En carcinomas suprarrenales la progresión de los síntomas es acelerada con un intervalo de 1 año entre la aparición de síntomas y el diagnóstico, existen excepciones sobre todo en niños quienes tienen mejor pronóstico. La virilización puede disminuir y la menstruación reanudarse tras la cirugía sobre todo en adenomas.
Los tumores malignos secretores de andrógenos, suelen ser de mayor tamaño al momento de la presentación que los tumores benignos (14 cm y 9 cm, respectivamente) y se presentan con valores séricos de testosterona 2,6 veces mayores en mujeres con tumores malignos14. Cuando cosecretan cortisol y andrógenos, los niveles de andrógenos no suelen ser tan elevados.
En el diagnóstico inicial de estos tumores se miden las concentraciones en suero de diferentes andrógenos: DHEAS, androstenediona y testosterona. Ningún andrógeno alterado único demostró tener suficiente especificidad y sensibilidad para diagnosticar estos tumores secretores11.
El mejor método para distinguir entre un tumor de origen ovárico versus suprarrenal es la tomografía computarizada (TC) como examen inicial, seguido de la resonancia magnética (RM) y la tomografía por emisión de positrones (PET), que permiten caracterizar la lesión y sugerir si el tumor es benigno o maligno2,14, así como valorar la presencia de metástasis5.
En cuanto a las características imagenológicas, se considera que el diámetro de la tumoración es predictivo de malignidad, de esta forma la mayoría de adenomas suprarrenales tienen menos de 4 cm de diámetro y los CAC son mayores a 4 cm al momento del diagnóstico5. Las lesiones malignas generalmente presentan mayor valor de atenuación o unidades de Hosunsfield (HU) y tumoraciones que presentan HU >20 y/o lavado de contraste <50% a los 10 minutos es más sospechoso de malignidad. Además, las masas tumorales pueden tener otros elementos sospechosos como heterogeneidad, bordes irregulares, calcificaciones, invasión de estructuras circundantes o adenomegalias15. En este caso no había elementos de sospecha de malignidad, excepto el tamaño del tumor.
La tomografía por emisión de positrones (PET) con fluorodesoxiglucosa (FDG) es buena para identificar tumores suprarrenales unilaterales con un mayor índice de sospecha de malignidad.
Lo llamativo de esta adolescente es que, desde el punto de vista anatómico, se trató de un tumor que no invadía estructuras vecinas dado el largo tiempo de evolución, como suele verse en los carcinomas. Sin embargo, la patología si tiene elementos claramente malignos o de mal pronóstico.
El tratamiento de elección para los tumores adrenales funcionantes es la cirugía. La adrenalectomía laparoscópica representa el tratamiento preferido en los CAC, pues acorta la estancia hospitalaria y reduce las complicaciones operatorias5.
La distinción entre tumores adrenocorticales benignos y malignos puede ser compleja, el único criterio diagnóstico definitivo de malignidad es la presencia de metástasis o la invasión local; en ausencia de estos hallazgos, la malignidad solo puede determinarse con el estudio anatomopatológico a través de los criterios microscópicos de Weiss16. Los cinco criterios utilizados en el sistema Weiss actualizado incluyen: >6 mitosis/50 campos de gran aumento (CGA), ≤25% de células tumorales claras en el citoplasma, mitosis anormales, necrosis e invasión capsular. Cada criterio se puntúa 0 cuando está ausente, o 2 para los dos primeros criterios y 1 para los tres últimos cuando está presente; el umbral de malignidad es una puntuación total ≥3. Para distinguir las neoplasias malignas se plantean criterios inmunohistoquímicos como el índice de proliferación Ki-67, pero los puntos de corte para lesiones benignas y malignas son muy variables. La sobreexpresión de P53, IGF-2 y ciclina E se encuentran en CAC pero no son suficientemente discriminatorias16. Se consideraron como factores con alto riesgo de recurrencia un Ki67 ≥10% y un índice mitótico ≥20/50 CGA17,18.
En adultos, en 2018 se implementó el sistema de estadificación TNM del Comité Conjunto Estadounidense sobre el Cáncer (AJCC) y la Unión para el Control Internacional del Cáncer (UICC)19.
Se ha utilizado un sistema algo diferente para niños, que identifica solo tres grupos distintos desde el punto de vista del pronóstico. En este sistema se incluyen menores de 20 años20. Los tres grupos son: 1. Tumores pequeños completamente resecados (200 g o menos): excelente pronóstico (supervivencia libre de eventos a 5 años del 91% en una serie de 228 niños). 2. Tumores grandes completamente resecados (más de 200 g), pronóstico intermedio (5 años, supervivencia libre de eventos 52% en la misma serie). 3. Enfermedad metastásica residual o a distancia, mal pronóstico6.
Conclusiones
Se presentó una paciente con carcinoma adrenal virilizante que comenzó con síntomas en la infancia. Este tipo de tumores tienen diferente comportamiento clínico entre pacientes adultos y pediátricos20. En niños se caracteriza por alta incidencia de virilización, elevados niveles de andrógenos y un curso clínico menos agresivo como vemos en este caso, a su vez parece ser más susceptible a la cirugía y otras modalidades terapéuticas. La resección quirúrgica completa mejora la supervivencia a largo plazo y la sobrevida a 5 años puede llegar al 100%9.
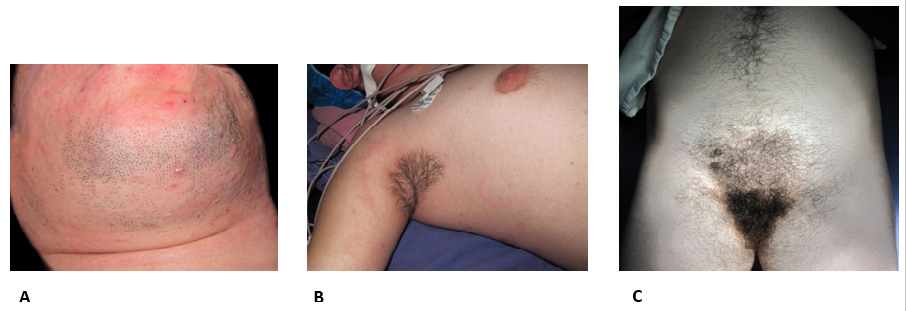
Figura 1: A) Acné y vello en mentón. B) Vello axilar y telarquia Tanner 1. C) Vello púbico y abdominal que evidencia el hirsutismo descrito.
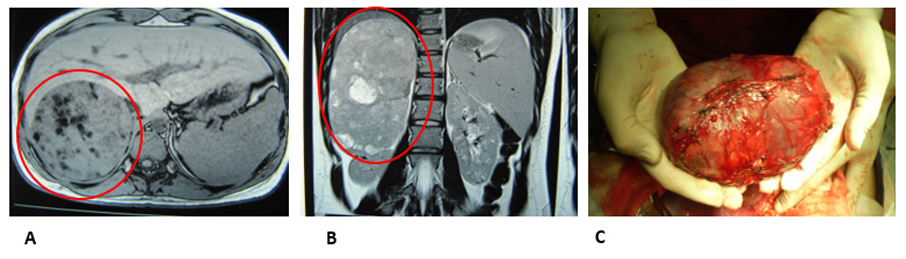
Figura 2: A) TC de abdomen (corte transversal): se observa masa suprarrenal derecha sólida, heterogénea, con sectores hipodensos y calcificaciones con bordes bien definidos, de 16x10x10 cm (círculo rojo). B) RM de abdomen (corte sagital): se observa la misma tumoración sólida (ovalo rojo) con realce inhomogéneo, contornos bien delimitados, algunas áreas sugestivas de necrosis. C) Pieza de resección: adrenalectomía con superficie externa lisa, completamente encapsulada.
Referencias
- Yang G, Dou J, Zhang X, Gu W, Lv Z, Du J, et al. Increased 3β‑hydroxysteroid dehydrogenase 2 and 17α‑hydroxylase activities in a virilized adolescent female with adrenal adenoma: A case report. Exp Ther Med. 2016; 11: 530-534.
- Navea C, Pavón I, Guijarro M, Merino M. Hyperandrogenism secondary to suprarenal tumor: Rare cause to take into account. Rev Int Androl. 2015; 13(4): 142-145.
- Ng L, Libertino J. Adrenocortical carcinoma: Diagnosis, evaluation and treatment. J Urol. 2003; 169: 5-11.
- Allolio B, Fassnacht M. Clinical review: Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab. 2006; 91(6): 2027-2037.
- Cordera F, Grant C, Van Heerden J, Thompson G, Young W. Androgensecreting adrenal tumors. Surgery. 2003; 134(6): 874-880.
- Michalkiewicz E, Sandrini R, Figueiredo B, Miranda E, Caran E, Oliveira-Filho A, et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. 2004; 22(5): 838-845.
- Fassnacht M, Johanssen S, Quinkler M, Bucsky P, Willenberg HS, et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: Proposal for a Revised TNM Classification. Cancer. 2009; 115(2): 243-250.
- Tong A, Jiang J, Wang F, Li C, Zhang Y, Wu X. Pure Androgen-Producing Adrenal Tumor: Clinical Features and Pathogenesis. Endocr Pract. 2017; 23(4): 399-407.
- Kerkhofs T, Ettaieb M, Verho even R, Kaspers G, Tissing W, Loeffen J, et al. Adrenocortical carcinoma in children: First population-based clinic pathological study with long-term follow-up. Oncol. 2014; 32: 2836-2844.
- Xiao XR, Ye LY, Shi LX, Cheng GF, Li YT, Zhouet BM. Diagnosis and treatment of adrenal tumor’s: a review of 35 years’ experience. Br J Urol. 1998; 82(2): 199-205.
- . Di Dalmazi G. Hyperandrogenism and Adrenocortical Tumors. Front Horm Res. 2019; 53:92-9.
- Icard P, Goudet P, Charpenay C, Andreassian B, Carnaille B, Chapuis Y, et al. Adrenocortical carcinomas: surgical trends and results of a 253 patient series from the French Association of Endocrine Surgeons study group. World J Surg. 2001; 25(7): 891-897.
- Stewart JN, Flageole H, Kavan P. A surgical approach to adrenocortical tumors in children: The mainstay of treatment. J Pediatr Surg. 2004; 39(5): 759-763.
- Moreno S, Montoya G, Armstrong J, Leteurtre E, Aubert S, Vantyghem M, et al. Profile and outcome of pure androgen-secreting adrenal tumors in women: experience of 21 cases. Surgery 2004; 136(6): 1192-1198.
- Szolar DH, Korobkin M, Reittner P, Berghold A, Bauernhofer T, Trummere H, et al. Adrenocortical carcinomas and adrenal pheochromocytomas: Mass and enhancement loss evaluation at delayed contrast-enhanced CT. Radiology. 2005; 234: 234(2): 479-485.
- Weiss Aubert S, Wacrenier A, Leroy X, Devos P, Carnaille B, Proye C, et al. system revisited: a clinic pathologic and immunohistochemically study of 49 adrenocortical tumors. Am J Surg Pathol. 2002; 26(12): 1612-1619.
- Fassnacht M, Dekkers O, Else T, Baudin E, Berruti A, de Krijger R, et al. European Society of Endocrinology Clinical Practice Guidelines on the Management of Adrenocortical Carcinoma in Adults, in Collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. 2018; 179(4): G1-G46.
- Beuschlein F, Weigel J, SaegerW, Kroiss M, Wild V, Daffara F, et al. Major Prognostic Role of Ki67 in localized adrenocortical carcinoma after complete resection. J Clin Endocrinol Metab. 2015; 100(3): 841-849.
- Phan AT, Grogan RH, Rohren E, Perrier ND. Adrenal Cortical Carcinoma. In: AJCC Cancer Staging Manual, 8th, Amin MB (Ed), Springer, New York. 2017: 911-912.
- Lam AK. Adrenocortical Carcinoma: Updates of Clinical and Pathological Features after Renewed World Health Organization Classification and Pathology Staging. Journal List Biomedicines. 2021; 9(2): 175.