Enfrentando la diabetes mitocondrial: Reporte de dos casos clínicos
Andrea Orellana T1,*, María José Valenzuela Pérez2, Mariana Ríos Soto1,3.
Mitochondrial Diabetes: Report of Two Clinical Cases
- Médico internista, Universidad de Valparaíso. Valparaíso, Chile.
- Diabetóloga y Nutrióloga. Médico Internista. Hospital Carlos Van Buren. Valparaíso, Chile.
- Médico cirujano, residencia medicina interna Universidad Valparaíso. Valparaíso, Chile.
*Correspondencia: Andrea Orellana T / a.orellanatoledo2292@gmail.com
Recibido: 22-05-2025.
Aceptado: 10-07-2025.
Resumen
Las enfermedades mitocondriales constituyen un grupo diverso de trastornos
genéticos que afectan principalmente órganos de alta exigencia metabólica,
como el sistema nervioso, el corazón, el hígado y el páncreas. Una manifestación
endocrinológica significativa es la diabetes mitocondrial, que representa
hasta el 3% de todos los casos de diabetes mellitus. Esta forma de diabetes
resulta de mutaciones en el ácido desoxirribonucleico mitocondrial (ADNmt)
que causan una secreción de insulina ineficiente. El diagnóstico de la diabetes
mitocondrial es complejo debido a su presentación heterogénea, que a
menudo comparte características clínicas tanto de diabetes tipo 1 y tipo 2.
Elementos clave para sospechar esta patología incluyen la afectación multiorgánica,
pérdida auditiva neurosensorial, y un índice de masa corporal bajo
a normal en adultos menores de 40 años. El diagnóstico definitivo se realiza
mediante la identificación de alteraciones en el ADNmt y su manejo requiere
un enfoque multidisciplinario. El control glicémico puede lograrse con insulina
y, en algunos casos, con metformina o inhibidores de la dipeptidil peptidasa
4 y análogos del péptido similar al glucagón tipo 1. La suplementación con
coenzima Q10, riboflavina y L-carnitina han demostrado ser beneficiosos para
mejorar la función mitocondrial y reducir los síntomas sistémicos. En el presente
artículo presentamos dos casos clínicos de diabetes mitocondrial, destacando
sus características diagnósticas, manejo clínico y evolución.
Palabras clave: Diabetes Mitocondrial; Hipoacusia; Enfermedades mitocondriales; MELAS; Síndrome de Kearns-Sayre.
Abstract
Mitochondrial diseases constitute a diverse group of genetic disorders that
primarily affect organs with high metabolic demands, such as the nervous
system, heart, liver, and pancreas. A significant endocrinological manifestation
is mitochondrial diabetes, which accounts for up to 3% of all diabetes mellitus
cases. This form of diabetes results from mutations in mitochondrial DNA (mtDNA) that lead to inefficient insulin secretion. The diagnosis of mitochondrial diabetes
is complex due to its heterogeneous presentation, often sharing clinical characteristics
with both type 1 and type 2 diabetes. Key elements for suspecting this condition include
multi-organ involvement, sensorineural hearing loss, and a low-to-normal body mass
index in adults under 40 years of age. Definitive diagnosis is made by identifying alterations
in mtDNA, and its management requires a multidisciplinary approach. Glycemic
control can be achieved with insulin and, in some cases, with metformin, dipeptidyl
peptidase-4 inhibitors, or glucagon-like peptide-1 receptor agonists. Supplementation
with coenzyme Q10, riboflavin, and L-carnitine has been shown to improve mitochondrial
function and reduce systemic symptoms. In this article, we present two clinical
cases of mitochondrial diabetes, highlighting their diagnostic characteristics, clinical
management, and evolution.
Keywords: Kearns-Sayre syndrome; Hearing Loss; MELAS; Mitochondrial Diabetes;
Mitochondrial Diseases.
Introducción
Las enfermedades mitocondriales constituyen un grupo diverso de trastornos genéticos que interfieren con la producción energética celular, afectando principalmente órganos de alta exigencia metabólica1.
Estas alteraciones dan lugar a manifestaciones multisistémicas que pueden variar en severidad y presentación.
Las mitocondrias convierten diversos sustratos de cofactores reducidos, derivados de los alimentos (como glucosa, ácidos grasos y cuerpos cetónicos), en trifosfato de adenosina (ATP), la principal fuente de energía celular. Las mutaciones en el ácido desoxirribonucleico mitocondrial (ADNmt) o en genes nucleares que codifican proteínas mitocondriales pueden interferir con este proceso, provocando una reducción en la producción de ATP y un aumento en el estrés oxidativo debido a especies reactivas de oxígeno2. Esta disfunción genera una amplia gama de manifestaciones que abarcan múltiples fenotipos.
Los síntomas neurológicos son comunes en este tipo de trastornos debido a la alta dependencia del sistema nervioso por el ATP generado por las mitocondrias. Estos síntomas pueden incluir accidentes cerebrovasculares “metabólicos”, migrañas, epilepsia, demencia, trastornos del movimiento, ataxia, pérdida auditiva neurosensorial y problemas de visión como ptosis, oftalmoplejía, atrofia óptica y retinitis pigmentosa. Dentro de las manifestaciones que comprenden al sistema nervioso periférico, los pacientes pueden experimentar ataxia sensorial, arreflexia, parestesias, miopatía, intolerancia al ejercicio y debilidad. Otros órganos que suelen estar afectados son el corazón, tracto gastrointestinal, hígado y el páncreas2,3.
La diabetes mitocondrial (DMmt) es una manifestación endocrinológica significativa. Ésta se debe a mutaciones en el ADNmt que causan una secreción de insulina ineficiente y subóptima debido a la disfunción en la fosforilación oxidativa. Se estima que representa hasta el 3% de todos los casos de diabetes mellitus4.
La DMmt incluye diversos fenotipos clínicos los cuales se resumen en la tabla 12,5.
El diagnóstico es desafiante debido a su presentación heterogénea, que a menudo comparte características con la diabetes tipo 1 y tipo 2. En algunos casos, la resistencia progresiva a la insulina exige un inicio más precoz de la insulinoterapia que en los pacientes con diabetes tipo 2. La presencia de afectación multiorgánica, especialmente pérdida auditiva neurosensorial, un índice de masa corporal bajo a normal y una presentación en menores de 40 años, son elementos clave para sospechar esta patología3,4. En este artículo, presentamos dos casos clínicos de diabetes mitocondrial, destacando sus características diagnósticas, manejo clínico y evolución.
Caso 1
Paciente femenina de 47 años, con antecedentes de miastenia gravis y diabetes mellitus de 5 años de evolución. Se derivó a estudio por historia de oftalmoparesia y pérdida de agudeza visual progresiva desde hace 30 años, asociada a debilidad fluctuante de extremidades inferiores e intolerancia al ejercicio. Además, la paciente reportó hipoacusia progresiva, retinopatía pigmentaria y oftalmoparesia bilateral, acompañada de parálisis facial periférica. Del estudio realizado, la resonancia magnética nuclear informó extensa leucoencefalopatía de sustancia blanca supra e infratentorial sugerente de patología metabólica. Electromiografía: Patrón miopático no inflamatorio. Ecocardiograma: Normal. Marcadores inmunológicos de diabetes: Negativo. Estudio genético molecular de deleciones en el ADNmt en sangre no detectó deleción.
Dado los antecedentes clínicos se descartó miastenia gravis y se diagnosticó mitocondriopatía sugerente de síndrome de Kearns-Sayre (SKS) y se inició tratamiento con suplementos de Arginina y coenzima Q10 para mejorar la función mitocondrial. En la última consulta, por glicemia de ayuno de 190 mg/dl y hemoglobina glicosilada (HbA1c) de 5.9% se indicó manejo no farmacológico por buen control metabólico.
Caso 2
Paciente femenina de 38 años, con antecedentes de epilepsia diagnosticada en la infancia en tratamiento con levetiracetam, hipoacusia severa bilateral y diabetes mellitus de 20 años de evolución en tratamiento con metformina y glibenclamida.
Presentó episodio de convulsión tónico-clónica que requirió manejo avanzado de la vía aérea y hospitalización en unidad de cuidados intensivos. En exámenes de ingreso solo destacaba hiperlactacidemia leve.
El análisis del líquido cefalorraquídeo mostró recuento normal de glóbulos blancos, proteínas y glucosa y lactato en 6,4 mmol/L (VN: 2,3 mmol/L). No se observaron levaduras capsuladas en la tinta china ni bacterias en la tinción de Gram. El cultivo fue negativo a las 48 horas y el VDRL resultó no reactivo.
El análisis de laboratorio complementario se resume en la tabla 2.
La tomografía computada (TC) de encéfalo no mostró lesiones, y se indicó tratamiento con ácido valproico al alta. Sin embargo, la paciente evolucionó con cefalea y dificultad para emitir lenguaje, razón por la cual consultó nuevamente. Se realizó un TC de encéfalo que informó extensa lesión hipodensa en territorio de la arteria cerebral media izquierda y atrofia supra e infratentorial. El estudio se complementó con angio resonancia que describió defecto de flujo en segmento C1 de carótida interna derecha y lesiones hiperintensas en DW1 y T2 en región temporoparietal izquierda y temporopolar derecha.
Tabla 1. Tipos de diabetes mitocondrial y sus principales características.
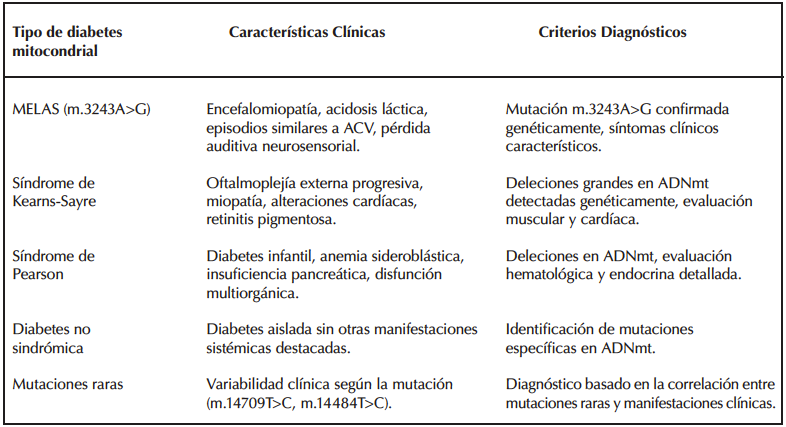
Tabla 2. Resultados de estudios complementarios realizados.s
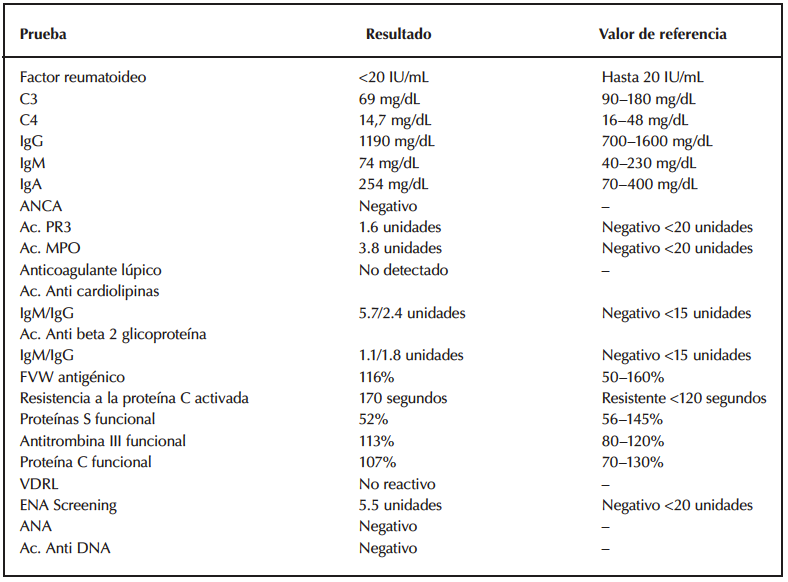
Ac: Anticuerpos; ANCA: Anticitoplasma de neutrófilos; PR3: Proteinasa 3; MPO: Mieloperoxidasa; FVW: Factor Von Willebrand.; ENA: Perfil de anticuerpos contra antígenos nucleares extraíbles; ANA: Anticuerpos antinucleares.
Frente a estos hallazgos, se sospechó síndrome de MELAS (encefalomiopatía mitocondrial, acidosis láctica y episodios similares a accidentes cerebrovasculares). Durante hospitalización cursó con mal control metabólico, siendo evaluada por diabetología, indicándose esquema basal bolo con insulina NPH y regular, logrando rápida compensación. Estudio genético molecular de MELAS en orina detectó la variante A3243G.
El manejo inicial incluyó dieta balanceada y ejercicio regular, además de insulina NPH a 0,5 UI/kg en dos dosis. Se inició tratamiento con lamotrigina para los episodios convulsivos y suplementación con L-arginina, L-carnitina y coenzima Q10. En el último control, presentó una HbA1c de 8,9% por falta de adherencia al tratamiento.
Discusión
La DMmt es una patología rara que debe ser considerada en pacientes con diabetes de inicio en la adultez joven acompañada de síntomas neuromusculares o multisistémicos6.
La complejidad de su diagnóstico radica en la coexistencia de moléculas de ADNmt normal y mutado dentro de una misma célula, tejido o individuo, fenómeno denominado heteroplasmia. Este equilibrio entre ADNmt sano y mutado varía entre tejidos y puede cambiar con el tiempo, lo que explica la variabilidad en la presentación clínica siendo necesaria una combinación de técnicas diagnósticas para lograr una identificación precisa7.
El diagnóstico definitivo se realiza mediante la identificación de alteraciones en el ADNmt, ya sean deleciones o variantes patogénicas, las cuales pueden ser detectadas en muestras de sangre, orina o biopsia muscular, de acuerdo con la sospecha clínica3.
Entre las distintas formas de DMmt, esta revisión se centra en el síndrome de Kearns-Sayre y MELAS, correspondientes a los casos clínicos analizados.
El SKS se caracteriza por la tríada clásica de oftalmoplejía externa progresiva, retinopatía pigmentaria y defectos de la conducción cardíaca, que normalmente se diagnostican antes de los 20 años, y se asocia a deleciones del ADNmt8,9. Para que estas deleciones sean detectadas se requiere un porcentaje mínimo de ADNmt mutado; si el nivel de heteroplasmia es muy bajo, la mutación puede no ser detectada1, como ocurrió en el primer caso clínico.
El síndrome MELAS se caracteriza por síntomas neurológicos, incluyendo episodios similares a los de un accidente cerebrovascular, y acidosis láctica. La variante genética más frecuentemente asociada a este síndrome es la mutación m.3243A>G del gen MT-TL1, identificada en el segundo caso clínico.
En 2012 se actualizaron sus criterios diagnósticos, que exigen al menos dos criterios clínicos (categoría A) y dos de laboratorio o genéticos (categoría B). La categoría A contempla síntomas clínicos y hallazgos de neuroimagen, como cefalea asociada a vómitos, convulsiones, hemiplejia, ceguera cortical y lesiones focales agudas en la corteza cerebral. La categoría B incluye lactato elevado en plasma o LCR, anomalías mitocondriales en biopsias musculares y una variante patogénica relacionada con MELAS identificada mediante pruebas genéticas.
Aunque estos criterios están bien definidos, persiste una gran variabilidad clínica, de imágenes y bioquímica entre los pacientes10.
El manejo específico de estas patologías incluye el uso de Coenzima Q10, L-Carnitina y Riboflavina que tienen como objetivo mejorar la función mitocondrial y reducir síntomas como la fatiga y debilidad muscular. Otros tratamientos como la dieta cetogénica, aún se encuentran en fase experimental4.
Las recomendaciones para el manejo farmacológico de la DMmt se basan principalmente en experiencia clínicia y extrapolación desde otras formas de diabetes. Aunque la metformina ha sido evitada por su asociación con acidosis láctica, estudios recientes muestran que este riesgo es bajo en ausencia de comorbilidades como insuficiencia renal, hepática o hipoxia. Puede considerarse en pacientes con función renal conservada, realizando ajuste de dosis con velocidad de filtración glomerular estimada entre 30–59 ml/ min/1,73 m2 y evitando su uso si ésta es <30 ml/min/1,73 m2, con monitoreo periódico de lactato.
Los inhibidores de la dipeptidil peptidasa-4 son bien tolerados y seguros, siendo una opción válida. Los análogos del receptor del pèptido similar al glucagón tipo 1 (aGLP- 1) y los inhibidores del cotransportador sodio-glucosa tipo 2, cuentan con beneficios cardiovasculares y renales demostrados en DM2 y podrían tener efectos favorables en DMmt, sin embargo, deben evitarse en enfermedad renal avanzada. Los aGLP-1 deben evitarse en pacientes con fenotipo gastrointestinal ya que podrían agravar sintomas digestivos3.
Los casos presentados ilustran la amplia variabilidad de la DMmt, atribuida en gran parte a la heteroplasmia, que puede generar diferencias tanto entre individuos como dentro de una misma familia. Un manejo multidisciplinario, flexible y adaptado a las necesidades de cada paciente, combinado con un diagnóstico temprano, resultan clave para mejorar significativamente el pronóstico y la calidad de vida de estos pacientes.
Referencias
- Wu B, Tao Y, Wu Q, Zou C, Liu X, Geng H. Mitochondrial diabetes presenting with spontaneous abortion and ketoacidosis onset: A case report and literature review. Medicine (Baltimore). 2024; 103(42): e40039. Doi: 10.1097/MD.0000000000040039. PMID: 39432645; PMCID: PMC11495764.
- El-Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. 2015; 116(1-2): 4-12. Doi: 10.1016/j.ymgme.2015.06.004. Epub 2015 Jun 15. PMID: 26095523.
- Karaa A, Goldstein A. The spectrum of clinical presentation, diagnosis, and management of mitochondrial forms of diabetes. Pediatr Diabetes. 2015; 16(1): 1-9. Doi: 10.1111/pedi.12223. Epub 2014 Oct 20. PMID: 25330715.
- Yeung RO, Al Jundi M, Gubbi S, Bompu ME, Sirrs S, Tarnopolsky M, Hannah-Shmouni F. Management of mitochondrial diabetes in the era of novel therapies. J Diabetes Complications. 2021; 35(1): 107584. Doi: 10.1016/j.jdiacomp.2020.107584. Epub 2020 Apr 13. PMID: 32331977; PMCID: PMC7554068.
- Chinnery PF. Primary Mitochondrial Disorders Overview. 2000 Jun 8 [updated 2021 Jul 29]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993–2024. PMID: 20301403.
- Alenezi AF, Almelahi MA, Fekih-Romdhana F, Jahrami HA. Delay in diagnosing a patient with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome who presented with status epilepticus and lactic acidosis: A case report. J Med Case Rep. 2022; 16(1): 361. Doi: 10.1186/s13256-022-03613- 2. PMID: 36210452; PMCID: PMC9549677.
- Cox BC, Pearson JY, Mandrekar J, Gavrilova RH. The clinical spectrum of MELAS and associated disorders across ages: A retrospective cohort study. Front Neurol. 2023; 14: 1298569. Doi: 10.3389/ fneur.2023.1298569. PMID: 38156086; PMCID: PMC10753009.
- Yee ML, Wong R, Datta M, Fazlo TN, Ebrahim MM, Mcnamara EC, De Jong G, Gilfillan C. Mitochondrial disease: An uncommon but important cause of diabetes mellitus. Endocrinol Diabetes Metab Case Rep. 2018; 2018: 18-0091. Doi: 10.1530/EDM-18-0091. PMID: 30306776; PMCID: PMC6169542.
- Grigalionienė K, Burnytė B, Balkelienė D, Ambrozaitytė L, Utkus A. Kearns-Sayre syndrome case. Novel 5,9 kb mtDNA deletion. Mol Genet Genomic Med. 2023; 11(1): e2059. Doi: 10.1002/mgg3.2059. Epub 2022 Oct 1. PMID: 36181358; PMCID: PMC9834195.
- Alves CAPF, Zandifar A, Peterson JT, Tara SZ, Ganetzky R, Viaene AN, Andronikou S, Falk MJ, Vossough A, Goldstein AC. MELAS: Phenotype Classification into Classic-versus-Atypical Presentations. AJNR Am J Neuroradiol. 2023; 44(5): 602-610. Doi: 10.3174/ajnr. A7837. Epub 2023 Apr 6. PMID: 37024306; PMCID: PMC10171385.