Función de los ácidos biliares en el control del metabolismo de la glucosa
BQ. Daniel Duran-Sandoval, PhD
Role of bile acids on glucose metabolism control
Departamento de Bioquímica Clínica e Inmunología, Facultad de Farmacia, Universidad de Concepción.
Correspondencia a:
Daniel Duran-Sandoval
Fax: 56-41-2207086
E-mail: dduran@ udec.cl
Recibido: 26 Septiembre de 2008
Aceptado: 20 Octubre de de 2008
Insulin resistance is the basis of several common diseases, such as type 2 diabetes, affecting millions people worldwide and satisfactory treatments are limited. Therefore, it is important to understand the molecular mechanisms underlying this condition and to find new and more effective therapies. Bile acids may actively participate in the control of metabolism. They derive from cholesterol, and function as natural ligands of nuclear and membrane receptors, regulating gene expression and controlling their own metabolism and that of glucose, including insulin response. Moreover, bile acids have been related to endoplasmic reticulum stress, a cellular response tightly associated to insulin resistance. These features give bile acids pharmacological properties with potential therapeutic use. Herein, we discuss the physiological role of bile acids on glucose metabolism, particularly on the regulation of the insulin response.
Key words: bile acids, insulina resistance.
La resistencia a la insulina (RI) es característica de la obesidad, la diabetes tipo 2 (DM2) y de los componentes del síndrome cardiometabólico, incluida la hipertensión y dislipidemia que, colectivamente, contribuyen al riesgo de enfermedades cardiovasculares. Las acciones metabólicas de la insulina en tejidos blanco clásicos (ej: músculo esquelético, adiposo y hepático), así como acciones en tejidos no clásicos (ej: cardiovascular), ayudan a explicar por qué la RI y la desregulación metabólica son centrales en la patogénesis del síndrome cardiometabólico y la patología cardiovascular1.
Aunque considerables progresos han sido realizados en el conocimiento de los mecanismos moleculares de la RI y la DM2, la disponibilidad de tratamientos satisfactorios aún es limitada, sirviendo más como medidas paliativas que curativas. De ahí la urgente necesidad de profundizar en los aspectos celulares que determinan la pérdida de respuesta a la insulina y el posterior desarrollo de complicaciones clínicas.
En la presente revisión se mostrarán una serie de datos que relacionan a los ácidos biliares con la respuesta a la insulina y la interesante posibilidad de su uso terapéutico en RI y DM2.
Mecanismos moleculares dependientes de ácidos biliares
Los ácidos biliares (ABs) son derivados del colesterol producidos en el hígado. Ellos ayudan a la solubilización, digestión y absorción de grasas y vitaminas liposolubles en el intestino. En el hombre, la síntesis de ABs a partir de colesterol permite la formación de 2 productos primarios principales: el ácido quenodesoxicólico (chenodesoxycholic acid, CDCA) y el ácido cólico (cholic acid, CA), quienes son conjugados con glicina o taurina y acumulados en la vesícula biliar hasta su utilización durante la digestión. Alternativamente, el ácido ursodesoxicólico (ursodeoxycholic acid; UDCA), un epímero hidrofílico de CDCA, es usado farmacológicamente como hepato-protector. Químicamente, los ABs son sintetizados por dos vías metabólicas: la vía neutra o “clásica”, que conduce a la formación de CDCA y CA, y la vía ácida o “alternativa” que conduce a la formación de CDCA principalmente. La diferencia entre ambas vías es que la vía ácida no es regulada negativamente por los propios ABs2. Gran parte de los ABs presentes en el lúmen intestinal son reabsorbidos, reactivados en el hígado y re-excretados en un ciclo conocido como circulación entero-hepática (CEH). La concentración intestinal de ABs aumenta durante al etapa post-prandial lo que favorece el aumento transitorio de los niveles plasmáticos de ABs en razón de su absorción intestinal. No obstante, los niveles plasmáticos de ABs son bajos (< 5-10 μM) debido a la alta capacidad de extracción del hígado3. Sin embargo, una alteración de los transportadores encargados de la movilización de ABs a través de la membrana plasmática, una inflamación hepática, un bloqueo de las vías biliares o el uso farma-cológico de ABs provocará elevación plasmática de ellos.
La identificación de receptores nucleares que responden a ABs y que son expresados en diferentes órganos, entre ellos el Receptor X Farnesoide (Farnesoid X Receptor; FXR)4-6, el Receptor X a Pregnano (Pregnane X Receptor; PXR)7 y el Receptor a la Vitamina D (Vitamin D Receptor; VDR)8, ha permitido comprender parcialmente el mecanismo molecular por el que los ABs regulan la expresión de genes y, por ende, el metabolismo. Ya sea como monómeros o formando heterodímeros con el receptor nuclear Receptor X a Retinoides (Retinoid X Receptor; RXR), los receptores nucleares activados por ABs son capaces de reconocer elementos de respuesta específicos situados en la región promotora de sus genes blanco modulando su expresión. Entre los diferentes ABs, el CDCA constituye el ligando natural endógeno más potente para FXR4-6; el ácido litocólico (litocholic acid, LCA) o su derivado acetilado lo son para PXR y VDR, respectivamente7,8. Sin embargo, diferentes estudios demuestran que los ABs pueden modular también la expresión de genes vía mecanismos que son independientes de la acción de receptores nucleares y que implicarían la activación de Proteinquinasas Activadas por Mitógenos (Mitogen-Activated Protein Kinases; MAPKs). Las MAPKs son enzimas conservadas evolutivamente y que conectan receptores de membrana a blancos reguladores clave en el interior de la célula. Los mamíferos expresan al menos 4 grupos de MAPKs: kinasas relacionadas a señales extracelulares (extracellular signal-related kinases; ERK-1/2), kinasas amino-terminales de Jun (Jun amino-terminal kinases; JNK-1/2/3), proteínas p38 (p38α/β/γ) y ERK5, que son activadas por kinasas de MAPKs específicas9. En efecto, los ABs libres o conjugados son capaces de activar ERK1/210-14, p3814,15 y JNK14-18 regulando efectos agudos en la célula. En este mismo contexto, gran interés ha suscitado el descubrimiento de la proteína TGR5 (Gpbar1, previamente descrita como BG37), un nuevo receptor de membrana acoplado a proteína G activado por ABs19,20. TGR5 se expresa en diversos tejidos del sistema entero-hepático y fuertemente en leucocitos, responde a formas libres y conjugadas de ABs aumentando los niveles de AMPc y es capaz de activar ERK1/219,20. Considerando las diversas vías de señales reguladas por los ABs, es que diversos laboratorios han desarrollado moléculas que permiten separar los mecanismos dependientes de aquellos independientes de receptores nucleares. Así, Malonney et al21, describió al GW4064, un agonista FXR no esteroidal de alta afinidad y que es utilizado en la actualidad como la “herramienta química” que permite evaluar los efectos dependientes de FXR. Además, una serie de moléculas naturales han sido descritas como ligandos para FXR. Entre ellas, Urizar y col22, describió que el esterol vegetal Z-guggulsterona actúa como un eficiente antagonista FXR suprimiendo los efectos FXR en diversos modelos celulares y también in vivo23-25. Sin embargo, algunos datos sugieren que Z-guggulsterona actuaría como un modulador selectivo de FXR23 (Figura 1).
Es evidente que existen una serie de vías por las que los ABs son capaces de regular la expresión de genes y, por ende, la función celular. Gracias a los avances de la biología molecular, una serie de modelos in vivo permiten comprender en la actualidad las funciones fisiológicas de estas vías de señales inducidas por ABs. Ello abre una interesante línea de investigación sobre la función que cumplen los ABs más allá del intestino y su potencial uso terapéutico.
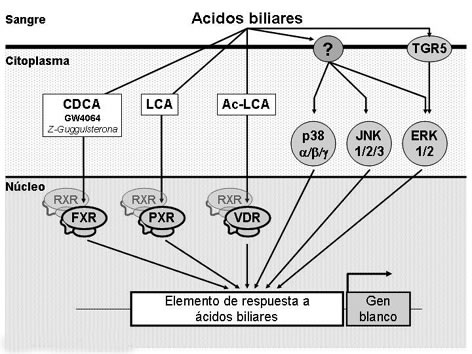
Figura 1. Mecanismos moleculares asociados a ácidos biliares. Los ácidos biliares (ABs) circulantes traspasan la membrana citoplasmática y actúan como ligandos de receptores nucleares, en particular CDCA como ligando de FXR, LCA como ligando de PXR y Ac-LCA como ligando de VDR. Además, GW4064 es descrito como un agonista sintético de FXR y Z-guggulsterona es utilizado como regulador negativo de la actividad FXR. Los receptores nucleares activados por ABs pueden formar heterodímeros con el receptor nuclear RXR y, así, unirse a la región promotora de genes blanco a través de elementos de respuesta a ABs, regulando su expresión. Alternativamente, los ABs pueden estimular moléculas de membrana y regular la actividad de MAPK tales como las familias p38, JNK y ERK. En particular, el receptor de membrana TGR5 estimula la vía ERK. Así, la regulación de las cascadas de señales MAPK permitirá también controlar la expresión de genes que posean en su región promotora elementos de respuesta a ABs. Para mayores detalles, referirse al texto principal.
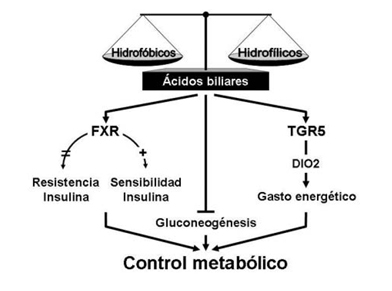
Figura 2. Efecto de los ácidos biliares sobre el control metabólico. La composición del pool de ácidos biliares (ABs) circulante fluctúa en concentración dependiendo del estado fisiopatológico del individuo de la presencia de ABs de tipo hidrofóbico o hidrofílico. Los ABs estimulan la actividad FXR y con ello la respuesta a la insulina. Sin embargo, la deficiencia de FXR se asocia a resistencia a la insulina. Además, los ABs regularían negativamente la gluconeogénesis. Por otra parte, vía TGR5, los ABs incrementan el gasto energético al favorecer la actividad DIO2, enzima clave en la síntesis intracelular de hormonas tiroideas activas. En conjunto, los ABs pueden controlar el metabolismo, un efecto que puede ser considerado en el contexto farmacológico.
ABs y metabolismo glucídico
Las observaciones sobre el efecto de los ABs en el balance energético y el metabolismo de azúcares han sido de gran interés. En efecto, la relación entre ABs y el metabolismo de carbohidratos nace desde las observaciones que de-muestran una disminución de la expresión hepática de FXR en varios modelos de diabetes y que la expresión de este gen es estimulada por glucosa y reprimida por insulina en hepatocitos primarios de rata in vitro26.
Además, la expresión hepática de FXR varía de acuerdo al estado nutricional, incrementando durante el ayuno y disminuyendo luego de una ingesta alimentaria27. Además, diferentes estudios sugieren que los ABs regularían negativamente la gluconeogénesis28,29. Sin embargo, los efectos in vivo sobre la glicemia que sean explicados por este mecanismo son limitados (Figura 2).
Actualmente, gran parte de la discusión se centra sobre el efecto de los ABs sobre la respuesta a la insulina. Recientemente, tres estudios independientes identifican un rol para FXR en la regulación de la sensibilidad a la insulina30-32. Ratones deficientes en FXR (FXR-/-) muestran disminución de la tolerancia a la glucosa y resistencia a la insulina. Usando el modelo de clamp euglicémico-hiperinsulinémico se ha confirmado que estos ratones presentan resistencia periférica a la insulina, que se refleja por disminución de la utilización periférica de la glucosa. En concordancia con esta observaciones, la cascada de señal de la insulina, evaluada por el nivel de fosforilación de la subunidad beta del receptor de insulina (IRβ) y de la proteinquinasa B (PKB/Akt) en respuesta a un bolus intraportal de insulina, se encuentra disminuida en tejidos periféricos sensibles a insulina tales como músculo esquelético y tejido adiposo blanco30,31. Sin embargo, los datos son discordantes respecto al nivel de sensibilidad hepática a la insulina en ratones FXR-/-. Algunos estudios observan una alteración de la inhibición mediada por insulina de la producción hepática de glucosa durante experimentos de clamp a baja dosis y de la señalización hepática de la insulina en ratones FXR-/-31,32. Por el contrario, la deficiencia en FXR también mostró estar asociada a normalidad de la sensibilidad y señalización hepática a la insulina27,30. Las razones de estas discrepancias no están claras, pero pueden deberse a diferencias en los fondos genéticos de los modelos FXR -/- utilizados (ratones C57Bl6/J31,32 versus C75Bl6/N27,30. Aún así, los ratones FXR-/- son un modelo de insulino-resistencia.
El efecto de la activación de FXR en modelos murinos de diabetes también ha sido estudiado. El tratamiento con GW4064 mejora significativamente la sensibilidad a la insulina en ratones db/db32 como también en ratones ob/ob30, ambos modelos de resistencia a la insulina. Resultados similares se obtuvieron cuando una forma constitutivamente activa de FXR, que no requiere unión de ligando, es sobre-expresada en ratones db/db32. Como FXR no se expresa en músculo esquelético, es probable que la alteración de la señalización de insulina en este tejido, que se observa en ratones FXR-/-, esté mediada por la elevación de los niveles circulantes de ácidos grasos libres31. Al respecto, datos recientes indican que FXR modula directamente la función del adipocito. Así, la activación de FXR mejora la señalización de insulina y la captación de glucosa inducida por insulina en adipocitos diferenciados desde la línea celular 3T3-L125,30 (Figura 2).
Otros estudios también indican que los ABs modulan la homeostasis de glucosa y el gasto energético de una manera independiente de FXR. En ratones alimentados con dietas suplementadas con CA, se previene la obesidad inducida por dieta al incrementar la expresión de la enzima yodotironina deyodinasa tipo 2 (DIO2). Este efecto se asocia a un incremento en la activación intracelular de hormonas tiroideas en tejido adiposo pardo murino y músculo esquelético humano33, que se correlaciona con el aumento del consumo de oxígeno en estas células. Un mecanismo probable es la regulación que ejerce TGR5 sobre la expresión de DIO2 estimulada por ABs33. Sin embargo, estudios in vivo han mostrado resultados contradictorios respecto a la función de TGR5 en la homeostasis energética34,35. Recientemente, resultados in vivo e in vitro sugieren que la retención de ABs hidrofóbicos confiere un estado de resistencia a la insulina en hígados colestásicos36. En particular, el ácido taurolitocólico 3-sulfato (Taurolithocholic acid-3 sulfate; TLCS) induce resistencia a la insulina por supresión de la fosforilación de la vía IRβ-PKB/Akt por un mecanismo dependiente de proteinquinasa C (PKC)36 (Figura 2).
Hasta ahora, no existirían datos en humanos sobre el efecto de ABs sobre la homeostasis de la glucosa. Sin embargo, se han obtenido interesantes resultados usando resinas secuestradoras de ABs, usadas clásicamente en pacientes hipercolesterolémicos. En un estudio transversal randomizado, doble ciego, la administración de colestira-mina mejora el control glicémico en pacientes con diabetes tipo 2 y dislipidemia37. Similares efectos benéficos sobre la homeostasis de glucosa fueron observados usando colestimida, otra resina de unión para ABs, en pacientes diabéticos tipo 238,39. Se requieren estudios adicionales para determinar si las acciones metabólicas de los secuestradores de ABs son producidos por mecanismos dependientes o independientes de las cascadas de señales mediadas por ABs en humanos.
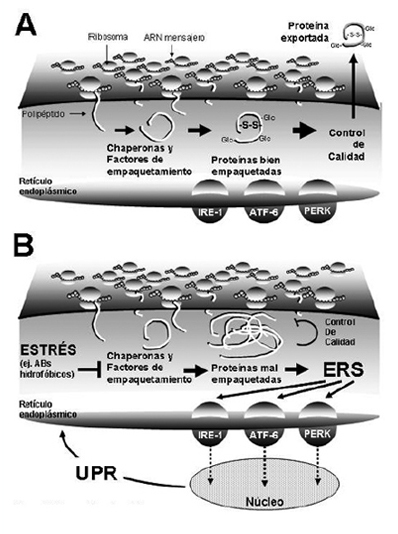
Figura 3. Ácidos biliares y estrés del retículo endoplásmico. A) Bajo condiciones de homeostasis celular, los ribosomas traducen los ARN mensajeros a polipéptidos que son transportados al interior del retículo endoplásmico. Estos polipéptidos son plegados y modificados químicamente a través de proteínas chaperonas y factores de empaquetamiento. Tras pasar el control de calidad en el retículo endoplásmico, las proteínas correctamente empaquetadas pueden ser exportadas para su utilización. B) Bajo condiciones de estrés, el proceso de empaquetamiento de proteínas falla, produciéndose una acumulación de proteínas mal empaquetadas en el lumen del retículo endoplásmico. Frente a este estrés del retículo endoplásmico (ERS), se estimulan cascadas de señales iniciadas por proteínas ancladas a la pared del retículo endoplásmico, incluidas IRE-1, ATF-6 y PERK. Estas proteínas transmitirán al núcleo la señal para la estimulación de la respuesta a proteínas no empaquetadas (UPR) que aliviará el ERS.
Estrés de retículo endoplásmico, respuesta a la insulina y ácido biliares
Estudios en la década pasada demostraron que la obesidad está asociada con inflamación y se estableció una relación entre respuesta inflamatoria y la acción anormal de la insulina40. Adicionalmente, diversas evidencias indican que las que las células β secretoras de insulina se pierden en DM1 y DM2 como resultado a respuestas estresantes reguladas por factores de trascripción y redes de genes claves41. Una serie de estudios sugieren que hay cascadas de señales pro-apoptóticas divergentes, corriente arriba en ambas formas de diabetes, dependientes de factores de transcripción aún no bien definidos42. Estas cascadas de señales primarias pueden converger en una única vía corriente abajo, tal como el estrés de retículo endoplásmico (endoplasmic reticulum stress; ERS), disfunción mitocon-drial, o producción de especies reactivas del oxígeno (ROS)41. ERS puede actuar también como un nexo entre obesidad y RI en el hígado y tejido adiposo, proponiendo la intrigante posibilidad que esta respuesta celular sea un mecanismo común para la falla de células β y el defecto de la señalización de la insulina en DM243. Así, diversas publicaciones demuestran que ERS está en la base tanto de la disfunción de células β pancreáticas como de la pérdida en tejidos periféricos de respuesta a la insulina42,44,45.
El retículo endoplásmico (endoplasmic reticulum; ER) es un organelo altamente dinámico con un rol central en la biosíntesis de lípidos y proteínas. El ER produce las proteínas de transmembrana y lípidos para la mayoría de los organelos y es responsable de la síntesis de la mayoría de las proteínas secretadas. El ER también tiene un rol importante en el almacenamiento y señalización dependiente de Ca2+. Debido a su habilidad de almacenar y secretar Ca2+, el ER controla un amplio rango de procesos celulares tales como la organogénesis, la actividad transcripcional, la respuesta al estrés y la apoptosis46.
La traducción de proteínas es desarrollada por los ribosomas en la superficie citosólica del ER47. En el lumen del ER estas cadenas son N-glicosiladas y empaquetadas en estructuras secundarias o terciarias que son estabilizadas por puentes disulfurados48. El ambiente oxidativo único del ER y las numerosas proteínas chaperonas presentes en el organelo son cruciales para el correcto empaquetamiento de proteínas y complejos proteicos49. La formación de puentes de disulfuro es catalizada por la proteindisulfuro isomerasa (protein disulfide isomerise; PDI)50. Otros factores de empaquetamiento presentes en le ER incluyen amino ácido cis-trans isomerasas, las chaperonas GRP94 y la proteína de unión a la cadena pesada de inmunoglobulinas (Ig heavy chain binding protein; BiP), enzimas para la N-glycosylation y las lectinas calnexina y calreticulina que específicamente acompañan los N-glicanos51, todos los que operan como complejos multiproteicos52. Mientras asistan al proceso de empaquetamiento, estas chaperonas y factores de empaquetamiento también retienen las proteínas cliente en el ER hasta que la proteína empaquetada reúna todas los estándares de control de calidad y salga del ER53-55.
El ER es exquisitamente sensible a alteraciones de la homeostasis. Como respuesta, las proteínas formadas en el ER pueden fallar en el logro de una correcta conformación debido a: pérdida de chaperonas o de la energía celular que promueva la interacciones chaperona-proteína, depleción de Ca2+, disrupción del estado redox, mutaciones proteicas que impiden un adecuado empaquetamiento y la reducción de los enlaces disulfuro. La acumulación de proteínas con fallas de empaquetamiento que se agregan en el lumen del ER causa estrés del ER (ERS) y la activación de señales de respuesta denominada UPR (Unfolded Protein Response; respuesta a proteínas no empaquetadas)56-59. El objetivo del UPR es aliviar el estrés del ER, restaurar la homeostasis del ER y prevenir la muerte celular. Para lograr estos objetivos la UPR coordina varias respuestas incluyendo: la disminución de la llegada de nuevas proteínas al ER previniendo así la alteración del empaquetamiento de nuevas proteínas y la sobrecarga del organelo; un incremento en la cantidad de chaperonas del ER aumentando así la capacidad de empaquetamiento del ER para tratar las proteínas mal empaquetadas; un aumento de la extrusión de las proteínas irreversiblemente mal empaquetadas desde el ER y su subsecuente degradación vía proteosoma; o iniciar la apoptosis si los pasos anteriores fallan. Debido a que estas respuestas dependen al menos en parte de la transcripción de genes de novo, las señales deben ser transmitidas desde el ER al núcleo indicando la necesidad urgente para la expresión de ARNm y proteínas relevantes. Esta señal es mediada por 3 proteínas de transmembrana del ER: la quinasa señal ER hacia núcleo requiriente de inositol 1 (inositol requiring ER-to-nucleus signal kinase; IRE-1), el factor de transcripción activante 6 (activating transcription factor; ATF-6) y la quinasa activada por ARN doble cadena similar a la quinasa de ER (double-stranded RNA-activated kinase (PKR)-like ER kinase; PERK). Estas proteínas se activan cuando proteínas no empaquetadas se acumulan en el lumen del ER y traducen esta información en señales que modulan la expresión de genes y proteínas clave, la UPR.
La acumulación de ABs hidrofóbicos en el hepatocito juega un rol clave en el daño del órgano en enfermedades hepáticas, tales como la colestasis. Se ha demostrado que el ácido desoxicólico (deoxycholic acid; DCA) y el ácido glicoquenodesoxicólico (glycochenodeoxycholic acid; GCDCA) inducen apoptosis in vitro e in vivo. Entre los mecanismos propuestos se incluye la alteración de la estructura y función de la membrana mitocondrial60, pero también la activación de genes pro-apoptóticos clave, incluyendo aquellos relacionados con ERS61. Así, diversos datos experimentales demuestran que la colestasis induce ERS en el hígado61. Actualmente, no está claro si el desarrollo de ERS hepático asociado a colestasis está relacionado con una alteración de la respuesta a la insulina. Sin embargo, estudios in vivo e in vitro sugieren que la retención de ABs hidrofóbicos confiere un estado de resistencia a la insulina en hígados colestásicos36. Es interesante notar además que pacientes con hígado graso no alcohólico, íntimamente relacionado a resistencia a la insulina, pueden cursar con signos clínicos de colestasis62,63. Sin embargo, se desconoce si la colestasia se correlaciona con pérdida de sensibilidad a la insulina.
Un estudio reciente demuestra que ABs hidrofílicos mejoran el ERS y la sensibilidad a la insulina. El tratamiento de ratones ob/ob con ácido tauroursodesoxicólico (taurour-sodeoxycholic acid; TUDCA), mejora la sensibilidad a la insulina y la glicemia sólo tras 10 días de tratamiento, a través de disminuir la producción hepática de glucosa y aumentar la remoción de glucosa en tejido graso y músculo esquelético64. Además, TUDCA alivia el ERS en hígado y tejido adiposo, disminuyendo la fosforilación de PERK and IRE1, reduciendo la fosforilación de c-Jun por JNK, y mejorando la señalización de insulina como lo demuestra el incremento de la fosforilación de IRβ, IRS-1 e IRS-2 en hígado y tejido graso64. Estos resultados sugieren la interesante hipótesis que los efectos biológicos de los ABs in vivo dependerán del tipo de ABs, hidrofílicos o hidrofóbicos, que prevalezcan en circulación.
Si bien hay evidencia sustancial que relaciona ERS con RI, actualmente no hay datos que evidencien el rol de ERS en tejidos humanos. Sin embargo, se ha observado un incremento en la expresión y grado de fosforilación de quinasas activadas por estrés, tales como p38 y JNK en tejido adiposo omental, pero no en tejido adiposo subcutáneo, aislado de mujeres obesas, comparadas con sujetos control65. Aunque no hay cambios evidentes en el nivel de fosforilación de IRS-1 estimulado por insulina en el tejido graso omental, la activación de quinasas que responden a estrés se correlaciona con la glicemia y con la RI observada en los pacientes estudiados65.
Proyecciones
Los ABs cumplen un rol metabólico importante que va más allá de su función en la digestión. Diversos estudios demuestran que los ABs ayudan a la integración metabólica regulando no sólo su propia síntesis sino también el metabolismo de azúcares y los requerimientos energéticos en diversos tejidos a través de señales intracelulares dependientes o independientes de receptores nucleares.
Si bien hacen falta mayores estudios en humanos, es interesante sugerir que trastornos hepáticos, como los observados en hígado graso no alcohólico, pudiesen estar asociados a una modificación en el metabolismo normal de los ABs con la consecuente acumulación de ABs de tipo hidrofóbico, lo que promoverá ERS en el hígado y otros tejidos con la consecuente pérdida de respuesta a la insulina. Sin embargo, ello permanece como una interesante hipótesis que requerirá se evaluada en futuros estudios
Evidenciando que los ABs son producidos por un órgano (el hígado) y que se encuentran presentes en la circulación, que su concentración varía en respuesta a los estímulos fisiopatológicos y, a su vez, promueven efectos biológicos a bajas concentraciones en diferentes órganos, mediados por receptores específicos, resulta interesante constatar que esas son características que describen a una hormona. Aunque considerar a los ABs como hormonas está lejos de ser aceptado, resulta prometedor sugerir al menos que estas moléculas pueden ser usadas farmacológicamente en patologías endocrinas que afectan ampliamente a nuestra población como lo son la RI y la DM2.
Agradecimientos
El desarrollo de este artículo fue financiado en parte por el proyecto DIUC 205.072.032-1.0, Dirección de Investigación y Facultad de Farmacia, Universidad de Concepción, Chile.
Referencias
- Permutt MA, Wasson J, Cox N. 2005. Genetic epidemiology of diabetes. J Clin Invest 115 (6): 1431-1439.
- Claudel T, Sturm E, Kuipers F, Staels B. 2004. The farnesoid X receptor: a novel drug target? Expert Opin Investig Drugs 13 (9): 1135-1148.
- Tolman KG, Rej R. 1999. Liver Function, in Tietz Textbook of Clinical Chemistry, B. C.A., Editor. Saunders W.B. Company: Phyladelphia. p. 1125-1177.
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. 1999. Identification of a nuclear receptor for bile acids. Science 284 (5418): 1362-1365.
- Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. 1999. Bile acids: natural ligands for an orphan nuclear receptor. Science 284 (5418): 1365-1368.
- Wang HJ Chen, Hollister K, Sowers LC, Forman BM. 1999. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 3 (5): 543-553.
- Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, et al. 2001. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A 98 (6): 3369-3374.
- Adachi R, Honma Y, Masuno H, Kawana K, Shimomura I, Yamada S, et al. 2005. Selective activation of vitamin D receptor by lithocholic acid acetate, a bile acid derivative. J Lipid Res 46 (1): 46-57.
- Chang L, Karin M. 2001. Mammalian MAP kinase signalling cascades. Nature 410 (6824): 37-40.
- Cheng K, Raufman JP. 2005. Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochem Pharmacol 70 (7): 1035-1047.
- Nakahara M, Fujii H, Maloney PR, Shimizu M, Sato R. 2002. Bile acids enhance low density lipoprotein receptor gene expression via a MAPK cascade-mediated stabilization of mRNA. J Biol Chem 277 (40): 37229-37234.
- Qiao D, Stratagouleas ED, Martínez JD. 2001. Activation and role of mitogen-activated protein kinases in deoxycholic acid-induced apoptosis. Carcinogenesis 22 (1): 35-41.
- Rao YP, Studer EJ, Stravitz RT, Gupta S, Qiao L, Dent P, et al. 2002. Activation of the Raf-1/MEK/ERK cascade by bile acids occurs via the epidermal growth factor receptor in primary rat hepatocytes. Hepatology 35 (2): 307-314.
- Yoon JH, Higuchi H, Werneburg NW, Kaufmann SH, Gores GJ. 2002. Bile acids induce cyclooxygenase-2 expression via the epidermal growth factor receptor in a human cholangiocarcinoma cell line. Gastroenterology 122 (4): 985-993.
- Bernt C, Vennegeerts T, Beuers U, Rust C. 2006. The human transcription factor AP-1 is a mediator of bile acid-induced liver cell apoptosis. Biochem Biophys Res Commun 340 (3): 800-806.
- De Fabiani E, Mitro N, Anzulovich AC, Pinelli A, Galli G, Crestani M. 2001. The negative effects of bile acids and tumor necrosis factor-alpha on the transcription of cholesterol 7alpha-hydroxylase gene (CYP7A1) converge to hepatic nuclear factor-4: a novel mechanism of feedback regulation of bile acid synthesis mediated by nuclear receptors. J Biol Chem 276 (33): 30708-30716.
- Gupta S, Stravitz RT, Dent P, Hylemon PB. 2001. Down-regulation of cholesterol 7alpha-hydroxylase (CYP7A1) gene expression by bile acids in primary rat hepatocytes is mediated by the c-Jun N-terminal kinase pathway. J Biol Chem 276 (19): 15816-15822.
- Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, et al. 2003. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev 17 (13): 1581-1591.
- Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. 2003. A G protein-coupled receptor responsive to bile acids. J Biol Chem 278 (11): 9435-9440.
- Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, et al. 2002. Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun. 298 (5): 714-9.
- Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, et al. 2000. Identification of a chemical tool for the orphan nuclear receptor FXR. J Med Chem 43 (16): 2971-2974.
- Urizar NL, Liverman AB, Dodds DT, Silva FV, Ordentlich P, Yan Y, et al. 2002. A natural product that lowers cholesterol as an antagonist ligand for FXR. Science 296 (5573): 1703-1706.
- Cui J, Huang L, Zhao A, Lew JL, Yu J, Sahoo S, et al. 2003. Guggulsterone is a farnesoid X receptor antagonist in coactivator association assays but acts to enhance transcription of bile salt export pump. J Biol Chem 278 (12): 10214-10220.
- Qin P, Borges-Marcucci LA, Evans MJ, Harnish DC. 2005. Bile acid signaling through FXR induces intracellular adhesion molecule-1 expression in mouse liver and human hepatocytes. Am J Physiol Gastrointest Liver Physiol 289 (2): G267-273.
- Rizzo G, Disante M, Mencarelli A, Renga B, Gioiello A, Pellicciari R, et al. 2006. The farnesoid X receptor promotes adipocyte differentiation and regulates adipose cell function in vivo. Mol Pharmacol 70 (4): 1164-1173.
- Durán-Sandoval D, Mautino G, Martin G, Percevault F, Barbier O, Fruchart JC, et al. 2004. Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes 53 (4): 890-898.
- Durán-Sandoval D, Cariou B, Percevault F, Hennuyer N, Grefhorst A, van Dijk TH, et al. 2005. The farnesoid X receptor modulates hepatic carbohydrate metabolism during the fasting-refeeding transition. J Biol Chem 280 (33): 29971-29979.
- De Fabiani E, Mitro N, Gilardi F, Caruso D, Galli G, Crestani M. 2003. Coordinated control of cholesterol catabolism to bile acids and of gluconeogenesis via a novel mechanism of transcription regulation linked to the fasted-to-fed cycle. J Biol Chem 278 (40): 39124-39132.
- Yamagata K, Daitoku H, Shimamoto Y, Matsuzaki H, Hirota K, Ishida J, et al. 2004. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J Biol Chem 279 (22): 23158-23165.
- Cariou B, van Harmelen K, Durán-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, et al. 2006. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem 281 (16): 11039-11049.
- Ma K, Saha PK, Chan L, Moore DD. 2006. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest 116 (4): 1102-1109.
- Zhang Y, Lee FY, Barrera G, Lee H, Vales C, González FJ, et al. 2006. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci USA 103 (4): 1006-1011.
- Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. 2006. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439 (7075): 484-489.
- Maruyama T, Tanaka K, Suzuki J, Miyoshi H, Harada N, Nakamura T, et al. 2006. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J Endocrinol 191 (1): 197-205.
- Vassileva G, Golovko A, Markowitz L, Abbondanzo SJ, Zeng M, Yang S, et al. 2006. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J 398 (3): 423-430.
- Mannack G, Graf D, Donner MM, Richter L, Gorg B, Vom Dahl S, et al. 2008. Taurolithocholic acid-3 sulfate impairs insulin signaling in cultured rat hepatocytes and perfused rat liver. Cell Physiol Biochem 21 (1-3): 137-150.
- Garg A, Grundy SM. 1994. Cholestyramine therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. A short-term, double-blind, crossover trial. Ann Intern Med 121 (6): 416-422.
- Kobayashi M, Ikegami H, Fujisawa T, Nojima K, Kawabata Y, Noso S, et al. 2007. Prevention and treatment of obesity, insulin resistance, and diabetes by bile acid-binding resin. Diabetes 56 (1): 239-247.
- Yamakawa T, Takano T, Utsunomiya H, Kadonosono, Okamura A. 2007. Effect of colestimide therapy for glycemic control in type 2 diabetes mellitus with hypercholesterolemia. Endocr J 54 (1): 53-58.
- Wellen KE, Hotamisligil GS. 2005. Inflammation, stress, and diabetes. J Clin Invest 115 (5): 1111-1119.
- Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. 2005. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 54 Suppl 2: S97-107.
- Eizirik, D.L., A.K. Cardozo, and M. Cnop. 2008. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 29(1): 42-61.
- Muoio, D.M. and C.B. Newgard. 2008. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 9 (3): 193-205.
- Sundar Rajan S, Srinivasan V, Balasubramanyam M, Tatu U. 2007. Endoplasmic reticulum (ER) stress & diabetes. Indian J Med Res 125 (3): 411-424.
- Tsiotra PC, Tsigos C. 2006. Stress, the endoplasmic reticulum, and insulin resistance. Ann N Y Acad Sci 1083: 63-76.
- Berridge MJ. 2002. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32 (5-6): 235-249.
- Adesnik M, Lande M, Martin T, Sabatini DD. 1976. Retention of mRNA on the endoplasmic reticulum membranes after in vivo disassembly of polysomes by an inhibitor of initiation. J Cell Biol 71 (1): 307-313.
- Kornfeld R Kornfeld S. 1985. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem 54: 631-664.
- Gething MJ, Sambrook J. 1992. Protein folding in the cell. Nature 355 (6355): 33-45.
- Bulleid NJ, Freedman RB. 1988. Defective co-translational formation of disulphide bonds in protein disulphide-isomerase-deficient microsomes. Nature 335 (6191): 649-651.
- van Anken E, Braakman I. 2005. Versatility of the endoplasmic reticulum protein folding factory. Crit Rev Biochem Mol Biol 40 (4): 191-228.
- Meunier L, Usherwood YK, Chung KT, Hendershot LM. 2002. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol Biol Cell 13 (12): 4456-4469.
- Brodsky JL. 2007. The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation). Biochem J 404(3): 353-363.
- Ellgaard L, Helenius A. 2003. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4 (3): 181-191.
- Schroder M, Kaufman RJ. 2005. The mammalian unfolded protein response. Annu Rev Biochem 74: 739-789.
- Patil C, Walter P. 2001. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol 13 (3): 349-355.
- Marciniak SJ, Ron D. 2006. Endoplasmic reticulum stress signaling in disease. Physiol Rev 86 (4): 1133-1149.
- Zhang K, Kaufman RJ. 2006. Protein folding in the endoplasmic reticulum and the unfolded protein response. Handb Exp Pharmacol (172): 69-91.
- Ron D, Walter P. 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8 (7): 519-529.
- Sola S, Brito MA, Brites D, Moura JJ, Rodrígues CM. 2002. Membrane structural changes support the involvement of mitochondria in the bile salt-induced apoptosis of rat hepatocytes. Clin Sci (Lond) 103 (5): 475-485.
- Tamaki N, Hatano E, Taura K, Tada M, Kodama Y, Nitta T, et al. 2008. CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am J Physiol Gastrointest Liver Physiol 294 (2): G498-505.
- Iorio R, Sepe A, Giannattasio A, Cirillo F, Vegnente A. 2005. Hypertransaminasemia in childhood as a marker of genetic liver disorders. J Gastroenterol 40 (8): 820-826.
- Sorrentino P, Tarantino G, Perrella A, Micheli P, Perrella O, Conca P. 2005. A clinical-morphological study on cholestatic presentation of nonalcoholic fatty liver disease. Dig Dis Sci 50 (6): 1130-1135.
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. 2006. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313 (5790): 1137-1140.
- Bashan N, Dorfman K, Tarnovscki T, Harman-Boehm I, Liberty IF, Bluher M, et al. 2007. Mitogen-activated protein kinases, inhibitory-kappaB kinase, and insulin signaling in human omental versus subcutaneous adipose tissue in obesity. Endocrinology 148 (6): 2955-2962.