Enfermedad de von Hippel Lindau en una familia chilena. Diagnóstico clínico y genético
Patricio Salman M.1,2, Nevenka Vucetich B.1,2 y José Manuel López M.1
Von Hippel Lindau disease. Report of an affected family
1Departamento de Endocrinología, Facultad de Medicina,
Pontificia Universidad Católica de Chile.
2Residente Endocrinología, Pontificia Universidad Católica de
Chile.
Correspondencia:
Patricio Salman M.
Departamento de Endocrinología,
Pontificia Universidad Católica de Chile.
E-mail: pasalman@med.puc.cl
Recibido: 24 de Noviembre de 2009
Aceptado: 11 de Diciembre de 2009
Von Hippel Lindau disease is a hereditary syndrome characterized by the appearance
of benign and malignant tumors in different organs. Its incidence is 1 case
per 36000 born alive. We report a family with the disease. The index case was a
male with a bilateral pheochromocytoma and cerebelar and retinal hemangioblastomas
that had a sudden death due to a cerebrovascular accident at the age
of 52 years. One sibling had central nervous system and retinal hemangioblastomas
and other was operated for an unilateral pheochromocytoma. Both siblings
had the R167Q VHL mutation of the syndrome. Other family members did not
have the mutation.
Key words: Enfermedad von Hippel Lindau, hemangioblastoma, feocromocitoma.
La enfermedad de von Hippel Lindau (VHL) es un síndrome neoplásico hereditario multisistémico, de transmisión autosómica dominante. Responde a una mutación germinal del gen supresor de tumores VHL, ubicado en el cromosoma 3. Se describe una incidencia de 1 caso por 36.000 nacidos vivos1.
Comunicamos el estudio de una familia chilena afectada por la enfermedad de VHL, creyendo de interés difundir el conocimiento sobre esta enfermedad y favorecer su temprano reconocimiento.
Caso clínico
Caso índice A. Varón de 29 años de edad, en quién, a raíz de un episodio de intensa cefalea asociada a hipertensión arterial, se sospechó aneurisma cerebral complicado; sin embargo, la arteriografía correspondiente no mostró alteraciones. Se mantuvo con hipertensión arterial leve y a los 47 años inicia episodio de mareos, inestabilidad de la marcha, sudoración, cefalea intensa y crisis hipertensiva.
Tratado con atenolol normaliza la presión arterial, con persistencia de la dificultad de equilibrio de la marcha.
A raíz de un hematocrito elevado (54%) se solicita TAC de abdomen que revela imágenes tumorales en ambas suprarrenales. Se certifica elevación de las catecolaminas urinarias con predominio de noradrenalina. El TAC de cerebro detectó un tumor cerebeloso, que se confirma con angiografía arterial y lo caracteriza con gran vascularización.
El efecto de masa se manifestaba como hidrocefalia. Considerando que el tumor cerebeloso estaba fuera del alcance quirúrgico, se realiza drenaje ventrículo peritoneal. Conjuntamente se efectúa suprarrenalectomía bilateral, confirmándose el diagnóstico de feocromocitoma bilateral, intraglandular, sin invasión de cápsulas ni vasos.
Se detectan además aneurismas retinales que es necesario fotocoagular en dos ocasiones. Al alta el paciente se mantiene normotenso, recibiendo cortisol oral y haciendo vida activa. A los 52 años de edad presentó cuadro agudo de cefalea intensa falleciendo abruptamente.
Su descendencia se componía de 4 hijos, dos hombres y dos mujeres.
Hija mayor. Caso B. Poco después de la muerte de su padre, a los 17 años, se realizó RM de cerebro que sólo destacó quiste pineal de 15 mm, con un discreto efecto de masa sobre el tercer ventrículo, sin interrumpir el paso de LCR, pero produciendo leve dilatación de los ventrículos laterales; la imagen cerebelosa era normal.
Reexaminada tres años después, su examen físico era normal, con normotensión y el fondo de ojo no mostraba lesiones vasculares. Repetida la RM de cerebro, se agrega a los hallazgos anteriores un pequeño angioma venoso en la fosa posterior. A partir de los 19 años se controla bianualmente con RM de cerebro, TAC abdominal, fondo de ojo y catecolaminas urinarias y metanefrinas, todas las cuales fueron normales. A los 25 años de edad se detecta un aneurisma retiniano que se coagula con láser. El estudio genético demuestra la mutación R167Q VHL (Universidad de Boston, USA). La paciente tuvo una hija cuyo estudio genético fue negativo.
Hijo menor. Caso C. Fue atendido por primera vez a los 14 años. Sin historia mórbida de importancia. Examen físico era normal y era normotenso. El TAC cerebral con especial estudio de fosa posterior fue normal. El TAC de abdomen reveló nódulo suprarrenal heterogéneo de 10,8 mm de diámetro; ambos riñones eran normales. La excreción urinaria de noradrenalina fue 135,8 μg/24 h y adrenalina fue 10,4 μg/24 h (VN suma de ambas < 100 μg/24 h). Cintigrama con MIBG demostró aumento de concentración del radiofármaco en la glándula suprarrenal derecha. Se realiza suprarrenalectomía derecha cuya biopsia certifica feocromocitoma. Se controla cada dos años con RM de cerebro, TAC de abdomen y examen retinal que siguen normales. El estudio genético resultó positivo para la mutación R167Q VHL (Universidad de Boston USA).
Hija menor. Caso D. Se ha mantenido asintomática y normotensa. El estudio inicial de fondo de ojo, catecolaminas urinarias, RM de cerebro y TAC de abdomen fueron normales. El estudio genético fue negativo para la mutación R167Q VHL (Universidad de Boston USA), por lo cual suspendió los controles.
Hijo mayor. Caso E. Siempre ha sido asintomático con exámenes iniciales normales. El estudio genético fue negativo para la mutación R167Q VHL (Universidad de Boston, USA).
En resumen, se presenta el caso de un paciente con un cuadro típico de VHL, fallecido a causa de sus complicaciones, y el estudio de sus cuatro hijos, dos de los cuales aparecen genéticamente heredando el defecto en el cromosoma 3.
A la fecha, los dos hijos afectados, se encuentran en control y sin evidencia de nuevos tumores asociados a esta enfermedad.
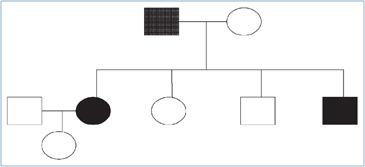 Figura 1. Árbol genealógico
del paciente
índice (caso A).
Figura 1. Árbol genealógico
del paciente
índice (caso A).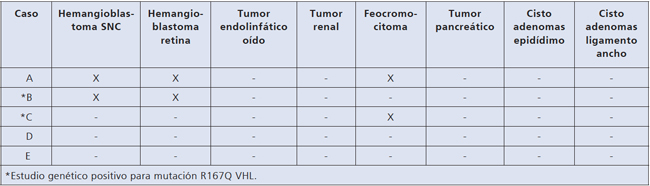 Figura 2. Esquema de patologías presentes en caso índice e hijos.
Figura 2. Esquema de patologías presentes en caso índice e hijos.
Discusión
La enfermedad de von Hippel Lindau es un síndrome neoplásico transmitido por herencia autosómica dominante y caracterizado por el desarrollo de tumores benignos y malignos en una variedad de órganos (OMIM n° 193300).
A los 65 años de edad tiene una penetrancia > 90%1.
Este síndrome resulta de una mutación del gen VHL en la línea germinal, el cual es un gen supresor de tumores localizado en el brazo corto del cromosoma 3 (3p25-26); está compuesto por 3 exones, codifica la proteína VHL y está ampliamente expresado en tejidos fetales y adultos.
El aislamiento del gen VHL se realizó en 19932-5. Según la hipótesis "two-hit" de Knudson, la formación de tumores surge cuando ambos alelos de VHL están inactivos, derivando en la pérdida de la capacidad supresora de tumores. La primera mutación inactivante es heredada, pero la segunda mutación es un evento somático6. Por otro lado, hay nuevos estudios que muestran otros posibles mecanismos para explicar esta enfermedad; así, se describe la proteína multifuncional vitronectina, que tiene actividad antiangiogénica y juega un importante rol en la regulación de la actividad de factores inducidos por hipoxia (HIF) tal como se expondrá más adelante. Mutaciones en la vitronectina, podrían afectar la represión de la angiogénesis y permitir así la aparición de hemangioblastomas. Además, la alteraciones de la vitronectina se correlacionan con formas más agresivas de VHL7,8. La proteína VHL influencia diversos procesos, incluyendo el control del ciclo celular, la estabilidad del mRNA y la regulación de la expresión de genes inducibles por hipoxia. La proteína VHL, en conjunto con otras proteínas, forma el complejo VCB-CUL2, el cual determina la proteolisis de diversas proteínas celulares. Cuando los niveles tisulares de oxígeno son normales, el complejo VCB-CUL2 se une a la subunidad α de los HIF 1 y 2, y de esta manera produce proteolisis de los HIF. Bajo condiciones de hipoxia las subunidades α no son degradables y así, tanto HIF1 como HIF2 activan la transcripción de un amplio repertorio de mRNA inducido por la hipoxia. Cuando el gen VHL está mutado, falla en su función de degradación de los HIF, lo que incrementa la expresión de factores angiogénicos, de crecimiento y mitogénicos, tales como el factor de crecimiento del endotelio vascular (VEGF), polipéptido β del factor de crecimiento derivado de las plaquetas (PDGFβ), eritropoyetina y el factor de transformación y crecimiento α (TGFα). Así, los HIF pueden estimular la angiogénesis, proceso que es crítico para la persistencia de los tumores asociados al síndrome de VHL3,4,9-11.
Aunque la mayor parte de los pacientes con VHL tienen una historia familiar positiva, sólo en el 20%8 de los casos se han descrito mutaciones de novo del gen y/o mosaicismos9.
El síndrome de VHL se clasifica clínicamente en los tipos 1 y 2, dependiendo si no tiene feocromocitoma (tipo 1) o sí lo presenta (tipo 2). A su vez, el VHL tipo 2 es clasificado en 3 categorías, denominadas VHL tipo 2A (feocromocitoma, hemangioblastomas del sistema nervioso central (SNC), hemangioblastoma retinal y tumores saco endolinfático), VHL tipo 2B (feocromocitoma, carcinoma renal, tumores pancreáticos, hemangioblastomas del SNC y retinal y tumores del saco endolinfático) y VHL tipo 2C (sólo feocromocitoma)10,12-14.
Otra enfermedad causada por inactivación del gen VHL es la policitemia de Chuvash, de carácter benigno y sin aparición de tumores10.
El diagnóstico de VHL se puede fundamentar con los criterios clínicos y/o en la positividad del test genético. En el caso de pacientes con historia familiar de VHL, hemangioblastomas de SNC, cáncer renal, feocromocitoma, quistes pancreáticos o tumores del saco endolinfático se sustentan los criterios para el diagnóstico de VHL. En ausencia de historia familiar (20%) se reúnen los criterios, de modo que el diagnóstico de vHL es sustentable si existen dos o más hemangioblastomas del SNC, o un hemangioblastoma y un tumor visceral asociado a VHL (con excepción de quistes renales y epididimarios)7.
Las manifestaciones clínicas y características del VHL son las siguientes:
1. Hemangioblastomas del SNC: son los tumores más comunes del síndrome, afectando al 60-80% de los pacientes. El promedio de edad de presentación es 33 años. Si bien son tumores benignos, constituyen la mayor causa de morbilidad. Se localizan a lo largo del eje craneoespinal, especialmente en la médula espinal y cerebelo, seguido por el tronco cerebral, raíces de nervios lumbosacros y región supratentorial. Los síntomas estarán relacionados con la localización del tumor, su tamaño, y la presencia de edema o quistes. Síntomas precoces son lumbalgia, cefalea, mareos y debilidad, entumecimiento o dolor en extremidades. La mejor evaluación de los hemangioblastomas extraretinales se consiguen con RM con contraste en fase T110.
2. Hemangioblastomas de retina: también llamados angiomas retinales, angiomatosis retinal o hemangiomas capilares yuxtapapilares; son de los tumores más comunes en el VHL (60%). Aparecen en la periferia retinal o cerca del nervio óptico, siendo a menudo multifocales y bilaterales (> 50%). La edad promedio de presentación es a los 25 años, aunque en un 5% pueden estar presentes antes de los 10 años de edad. Los angiomas retinales son asintomáticos en los estadios iniciales, pero pueden conducir precozmente a pérdida parcial o total de la visión. La oftalmoscopía con dilatación del iris permite la identificación de la mayoría de los tumores retinales, y ante dudas se puede completar el estudio con angiografía retinal con fluoroceína. Histológicamente son idénticos a los hemangioblastomas del SNC15,16.
3. Tumores del saco endolinfático: son tumores de rara presentación en la población general, pero frecuentemente asociados a VHL (11%). Dependiendo de la localización, los pacientes pueden presentar pérdida parcial o total de la audición, tinnitus, sensación de inestabilidad y paresia facial. Estos tumores son muy vascularizados y a menudo erosionan o se expanden hacia el hueso subyacente. Pueden recurrir, pero no dan metástasis. El diagnóstico y localización se realiza con RM o TAC con contraste15,16.
4. Quistes y carcinoma de células renales: es la principal neoplasia maligna en VHL, y es la primera causa de cáncer renal genético (24-45%). Los quistes renales hacen subir el porcentaje de lesiones renales a 60%. La edad promedio de presentación es 39 años (25 años más temprano que el carcinoma renal esporádico), y tienden a crecer más lento. Las lesiones renales son a menudo múltiples y bilaterales. El tipo histológico de cáncer renal asociado a VHL es siempre de células claras. En el diagnóstico y seguimiento de estas lesiones se recomienda el uso de RM sobre el TAC15-17.
5. Feocromocitoma: se presentan en un 10-20% de los pacientes con VHL. La edad de aparición es después de los 30 años (más temprano que en los casos esporádicos); pueden ser múltiples y bilaterales, y en algunos individuos son la única manifestación del síndrome (VHL tipo 2C). También se han descrito paragangliomas extra-adrenales (10%). Funcionalmente los feocromocitomas del VHL sólo producen noradrenalina lo que implica medir las normetanefrinas dentro del proceso diagnóstico. La localización se puede conseguir con RM o TAC de abdomen con contraste; no obstante, para tumores cromafines extraadrenales pueden necesitarse técnicas adicionales (MIBG, 18F-DOPA PET). Aunque a menudo el feocromocitoma en el VHL es asintomático, su comportamiento es impredecible: lesiones inactivas pueden bruscamente tornarse agresivas, o lesiones benignas pueden malignizarse; con todo, sólo el 5% de estos feocromocitomas son malignos11,15,16,18.
6. Quistes y tumores pancreáticos neuroendocrinos: estos tumores sólidos se presentan en 8-17% de los pacientes con VHL, mientras que los quistes y cistoadenomas serosos se evidencian en el 17-56%. La edad promedio de presentación de los tumores neuroendocrinos sólidos es 35 años, y la de los quistes pancreáticos, 37 años. En general son asintomáticos. Algunos de los tumores son múltiples y malignos, y la mayor parte no son funcionantes. El diagnóstico puede ser sospechado en un TAC con contraste y puede confirmarse con RM15,16,19.
7. Cistoadenomas epipidimarios: son vistos en el 25-60% de los varones con VHL y pueden ser múltiples y bilaterales. Son benignos, asintomáticos y típicamente aparecen en la adolescencia. El diagnóstico puede ser hecho por palpación y confirmado por criterios ultrasonográficos15.
8. Cistoadenomas del ligamento ancho: estos son raramente comunicados y muchas veces no reconocidos en muchas mujeres con VHL. La edad de presentación y la real frecuencia de estos cistoadenomas es desconocida. Son generalmente asintomáticos y pueden ser ubicados con TAC y ultrasonografía15. En el caso específico de nuestra familia el síndrome de VHL heredado del padre comprometió al 50% de sus hijos. La localizaciones detectadas fueron en el padre hemangioblastoma de SNC y retina y feocromocitoma, en la hija mayor hemangioblastoma de SNC y retina y en el hijo menor feocromocitoma.
El tratamiento del VHL va a depender del o de los órganos comprometidos y el tipo de lesiones (benignas o malignas) y se resume de la siguiente manera: a) la mayor parte de los hemangioblastomas pueden ser completamente resecados quirúrgicamente en forma exitosa y segura, y el momento de hacerlo corresponde al inicio de la sintomatología. En algunos centros se ha utilizado la embolización preoperatorio; b) Los hemangioblastomas retinales responden la mayoría de las veces a fotocoagulación con láser o crioterapia; c) los tumores del saco endolinfático se operan y ello es curativo cuando la lesión se reseca completamente, y usualmente se preserva la capacidad auditiva; d) El cáncer de células renales es de resorte quirúrgico aunque está limitado por el tamaño del tumor; en casos de tumores muy pequeños se han utilizado tratamientos con radiofrecuencia percutánea o crioablación y drogas; e) el tratamiento del feocromocitoma es quirúrgico (preferentemente laparoscópico) requiriendo control y manejo preoperatorio farmacológico adecuado; f) los tumores pancreáticos deben operarse con las limitaciones propias de la localización y el tamaño; a este respecto, Libutti y colegas recomiendan los siguientes criterios para indicar la resección: 1) sin evidencia metastásica, 2) tamaño tumoral > 3 cm en el cuerpo o cola del páncreas, o > 2 cm en la cabeza del páncreas; 3) paciente que debe ser sometido a laparotomía por otra patología. También se ha extendido la indicación quirúrgica a pacientes con tumores pancreáticos, incluso si existen metástasis a linfonodos regionales; g) el tratamiento de los cistoadenomas epididimarios es conservador y su extirpación se reserva en el raro caso de ser sintomático; lo mismo ocurre con los cistoadenomas del ligamento ancho10,16.
Se ha estudiado la terapia anti-angiogénica para diversos tumores relacionados con la enfermedad de VHL.
Recientemente se publicó un caso clínico donde se utilizó el inhibidor de la tirosina-kinasa Sunitinib en un paciente con VHL, que presentaba un feocromocitoma maligno metastásico; fue tratado por 6 meses, con notable mejoría respecto de la hipertensión, baja de peso, dolor y con reducción de las metanefrinas. Este fármaco fue aprobado por la Food and Drug Administration de EE.UU. el año 2006 para el tratamiento del cáncer renal y los GIST. Este fármaco está en pleno proceso de estudio y validación para este tipo de pacientes20.
Finalmente, ante una enfermedad tan compleja y con tantas posibilidades de complicaciones debe enfatizarse la necesidad de mantener un alto nivel de sospecha por parte de los médicos generales que recibirán inicialmente al paciente. La búsqueda del componente hereditario, cuando positivo, debe acentuar esta sospecha. El reconocimiento precoz del síndrome puede ayudar al paciente y favorecer la identificación de otros miembros comprometidos de la familia. Así es plenamente aconsejable efectuar examen de fondo de ojo en pacientes con hipertensión arterial y también en los hipertensos buscar otras localizaciones tumorales cuando se diagnostica una de ellas.
Referencias
- Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT. 1990. Clinical features and natural history of von Hippel-Lindau disease QJM; 77: 1151-1163.
- Latif F, Tory K, Gnarra J, Yao M, Duh F, Orcutt M, et al. 1993. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260; n° 5112, 1317-1320.
- Kim WY, Kaelin WG. 2004. Role of VHL gene mutation in human cancer. J Clin Oncol 22: 4991-5004.
- Iliopoulos O, Kibel A, Gray S, Kaelin Jr WG 1995. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1: 822-826.
- Clark PE, Cookson MS. 2008. The von Hippel Lindau gene. Cancer; 113 (7 suppl): 1768-1778.
- Knudson AG Jr. 1971. Mutation and cancer: statistical study of retinoblastoma. Proc Nat Acad Sci USA: vol 68, n° 4, 820-823.
- Turturro F. 2009. Beyond the Knudson´s hypothesis in von Hippel-Lidau (VHL) disease-proposing vitronectin as a "gene modifier". J Mol Med; 87: 591-593.
- Huang JH, Lin CM, Cheng YC, Hung KL, Chien CC, Chen SK. 2009. A vitronectin M381T Polymorphism increases risk of hemangioblastoma in patients with VHL gene defect. J Mol Med 87: 613-622.
- Kaelin WG Jr. 2007. von Hippel-Lindau disease. Annu Rev Pathol Mech Dis 2: 145-173.
- Shuin T, Yamazaki I, Tamura K, Kamada M, Ashida S. 2004. Recent advances in ideas on the molecular pathology and clinical aspects of von Hippel-Lindau disease. Int J Clin Oncol 9: 283-287.
- Woodward ER, Maher ER. 2006. von Hippel-Lindau disease endocrine tumour susceptibility. Endocrine-Related Cancer 13: 415-425.
- Neumann HPH, Wiestler OD. 1991. Clustering of features of von Hippel-Lindau disease: evidence of a complex genetic locus. Lancet 337: 1052-1054.
- Brauch H, Kishida T, Glavac D, Chen F, Pausch F, Höfler H, et al. 1995. von Hippel-Lindau (VHL) disease with pheochromocytoma in the Black Forest region in Germany: evidence for a founder effect. Hum Genet 95: 551-556.
- Chen F, Kishida T, Masahiro Y, Hustad T, Glavac D, Dean D, et al. 1995. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlation with phenotype. Hum Mutat 5:66-75.
- Sano T, Horiguchi H. 2003. von Hippel-Lindau disease. Microsc Res Tech 60: 159-164.
- Lonser RR, Glenn GM, Walther M, Chew EY, Libutti ST,
Linehan WM, et al. 2003. von Hippel-Lindau disease. Lancet
361: 2059-2067. - 17. Opocher G, Conton P, Schiavi F, Macino B, Mantero F. 2005. Pheochromocytoma in von Hippel-Lindau disease and neurofibromatosis type 1. Familial Cancer 4: 13-16.
- Meister M, Choyke P, Anderson C, Patel U. 2009. Radiological evaluation, management, and surveillance of renal masses in von Hippel-Lindau disease. Clinical Radiology 64: 589-600.
- Ros LH, García AI, Torres GM, Ros PR. 1997. Magnetic resonance imaging evaluation of a case of von Hippel-Lindau disease. Eur Radiol 7: 1282-1284.
- Jiménez C, Canabillas M, Santarpia L, Jonasch E, Kyle K, Lano E, et al. 2009. Use of the tyrosine kinase inhibitor Sunitinib in a patient with von Hippel-Lindau disease: targeting angiogenic factors in pheochromocytoma and other von Hippel-Lindau disease disease-related tumors. J Clin Endocrinol Metab 94: 386-391.