Tumor ovárico de Células de Leydig en una niña de 7 años que se presentó como Pubarquia Precoz con elevación de 17 Hidroxiprogesterona
Mónica Arancibia1, Hernán García F2, Alejandro Martínez1, Paulina Baquedano3, Cristián García B 4, Hernán García B1.
Ovarian tumor of Leydig cells in a 7-year-old girl who presented as precocious pubarche with elevation of 17 Hydroxyprogesterone
1Endocrinología Pediátrica. Departamento de Pediatría. Pontificia Universidad Católica de Chile.
2Alumno de Medicina Séptimo año. Pontificia Universidad Católica de Chile.
3Urología Pediátrica. Departamento de Urología. Pontificia Universidad Católica de Chile.
4Departamento de Radiología. Pontificia Universidad Católica de Chile.
Correspondencia: Hernán García Bruce Dirección postal: Diagonal Paraguay 363, 8º piso. División de Pediatría. teléfono: 569 9824 5581 oficina: 2 2354 3402 Correo electrónico: hgarciabr@uc.cl.
Recibido: 04-08-2018
Aceptado: 20-11-2018
Abstract: Clinical case: a girl of 7 ½ years who consulted for early pubarche without thelark, with a percentile size of 75 for a genetic target size in the 10th percentile, overweight with a 90th percentile BMI, and normal blood pressure. The biochemical study showed high levels of androgens: testosterone: 7.2 ng/dL, androstenedione of 5.1 ng / ml, 17OHP: 15 ng / dL with low normal DHEAS (0.26 ug/ml), Plasma Renin Activity normal low: 0.22 ng/mL/h. Initial imaging study showed a bone age of 10 years 6 months and normal abdominal and pelvic ultrasound. Molecular study showed no pathogenic variants in the CYP21A2 gene (21 Hydroxylase). With a probable diagnosis of non-classical congenital adrenal hyperplasia (HSRNC) and no known mutation, he started treatment with hydrocortisone (12 mg/m2). At 8.7 years, pubertal development begins and braking begins with LHRH analogues, which are administered for 18 months. Despite the treatment, signs of virilization and elevation of androgens (testosterone up to 130 ng/ml) are progressively accentuated, which do not diminish when trying different corticosteroid schemes. MRI of the abdomen and pelvis shows the normal adrenal glands and a solid nodular image of 2.1 x 1.6 cm in the right ovary (Figure 2), later demonstrated with pelvic ultrasound (Figure 2). Right laparoscopic oophorectomy was performed, whose biopsy demonstrated a Leydig cell tumor. One month after surgery, all androgenic levels were normalized, so the gradual suspension of corticosteroids began. Conclusion: Although HSRNC is the most frequent pathological cause of early pubarche, when it is associated with progressive clinical and biochemical hyperandrogenism despite adequate treatment and without pathogenic variants in the CYP21A2 gene, even with high levels of 17OHP, other causes should be considered, specifically, androgen producing tumors.
Key words: Adrenal Hyperplasia; Hirsutism; Leydig cells; Ovarian tumor.
Resumen: Caso clínico: niña de 7½ años que consulta por pubarquia precoz sin telarquia, con talla en percentil 75 para una talla objetivo genético en percentil 10, sobrepeso con IMC percentil 90 y presión arterial normal. El estudio bioquímico mostró niveles elevados de andrógenos: testosterona: 7,2 ng/dL, androstenediona de 5,1 ng/ml, 17OHP: 15 ng/dL con DHEAS normal baja (0,26 ug/ml), Actividad de Renina Plasmática normal baja: 0.22 ng/ mL/h. Estudio de imágenes inicial mostró una edad ósea de 10 años 6 meses y ecografía abdominal y pelviana normales. Estudio molecular no mostró variantes patogénicas en el gen CYP21A2 (21 Hidroxilasa). Con diagnosticó probable de hiperplasia suprarrenal congénita no clásica (HSRNC) y sin mutación conocida,inició el tratamiento con hidrocortisona (12 mg/m2). A los 8.7 años comienza desarrollo puberal y se inicia frenación con análogos de LHRH, los cuales se administran por 18 meses. A pesar del tratamiento se acentúan progresivamente los signos de virilización y hayelevación de los andrógenos (testosterona hasta 130 ng/ml), que no disminuyen intentando diferentes esquemas de corticoides. Se realiza RM de abdomen y pelvis que muestra las glándulas suprarrenales normales y una imagen nodular sólida de 2.1 x 1.6 cm en el ovario derecho (Figura 2), demostrada posteriormente con Ecografía pelviana (Figura 2). Se realiza ooforectomía derecha por vía laparoscópica, cuya biopsia demostró un tumor de células de Leydig. Un mes después de la cirugía, se normalizan todos los niveles androgénicos por lo que se inició la suspensión gradual de los corticoides. Conclusión: Aunque la HSRNC es la causa patológica más frecuente de la pubarquia precoz, cuando se asocia con un hiperandrogenismo clínico y bioquímico progresivo a pesar de un tratamiento adecuado y sin variantes patógenicas en el gen CYP21A2, incluso con niveles elevados de 17OHP, otras causas deben ser consideradas, específicamente tumores productores de andrógenos.
Palabras clave: Células Leydig, Hirsutismo, Hiperplasia Suprarrenal; Tumor ovárico.
Objetivo: Reportar el caso clínico de una niña de 7½ años que consultó por pubarquia precoz y signos de virilización progresiva debido a hiperandrogenismo producido por un tumor ovárico de células de Leydig, inicialmente diagnosticada como HSRNC, por elevación de 17OHP.
Caso Clínico
Paciente de sexo femenino que consulta a los 7 años 10 meses por pubarquia precoz (PP), sin antecedentes mórbidos de importancia. Producto de embarazo fisiológico, parto normal, recién nacido de término de 3.740 g de peso y 50 cm de longitud. Desarrollo psicomotor normal. Madre de 43 años, sana, menarquia a los 11 años, Talla 153 cm. Padre 45 años, sano, desarrollo puberal normal, Talla 168 cm. (Talla objetivo genético: p10). Dos hermanos (varón y mujer de 11 y 5 años) sanos.
Al examen se confirma vello púbico Tanner 3, acné facial moderado, olor apocrino y piel grasa, sin aumento de tamaño del clítoris. Hirsutismo con puntaje de Ferriman 8, Mamas Tanner I. Talla de 132 cm (p75); Índice de Masa Corporal 19,2 kg/m2 (p 90) y presión arterial 100/70 mmHg (p63).
El estudio bioquímico mostró niveles elevados de Testosterona, Androstenediona y 17OHP, con DHEAS normal (Tabla 1). Radiografía (Rx) de mano mostró una edad ósea de 10 años 6 meses para 8 años y ecotografía de abdomen y pelvis inicial mostraron útero y ovarios prepuberales sin alteraciones. El estudio molecular de amplificación del gen activo para HSRNC (CYP21A2), pseudo-gen y genes híbridos; y su posterior secuenciación, no mostró variantes patogénicas. Basado en el cuadro clínico se diagnosticó HSRCN sin mutación demostrable y se inició tratamiento con hidrocortisona (12 mg/ m2/día) con escasos resultados clínicos y bioquímicos (Tabla 1).
A los 8,7 años se agrega desarrollo mamario, confirmándose inicio de pubertad central temprana mediante Test de estímulo con LHRH, (pico de LH de 8 mIU/L (eje activado: LH > 5 mUI/ mL). Por importante avance de edad ósea se inicia frenación pubertal con análogo de LHRH (Triptorelina 11.25 mg cada 3 meses), el cual se mantuvo por 18 meses.
A pesar del tratamiento, los signos de virilización se acentuaron progresivamente, con aumento del hirsutismo, voz de tono más grave e hipertrofia del clítoris, todo esto asociado a elevación de las concentración de andrógenos: Testosterona (T) hasta 133 ng/mL y Androstenediona (A) > a 10 ng/ml (Tabla 1).
A pesar que se intentó diferentes esquemas terapéuticos con hidrocortisona en dosis más altas (hasta 22 mg/kg/día), luego prednisona y posteriormente dexametasona, no se reducen las concentraciones de T ni A elevadas, patrón característico de hiperandrogenismo independiente de ACTH. Se mantuvo elevado.
Se realiza RM de abdomen y pelvis que muestra las glándulas suprarrenales normales y una imagen nodular sólida de 2.1x1.6 cm en el ovario derecho (Figura 1), demostrada posteriormente con ecografía pelviana (Figura 1).
Con diagnóstico de tumor ovárico productor de andrógenos, posiblemente tumor de Células de Sertoli-Leydig o Tumor de células de la Granulosa, a los 10 años 4 meses se realiza laparoscopia que confirma tumor en el ovario derecho por lo que se realiza salpingooforectomía (Figura 2). El ovario izquierdo era de aspecto normal. El estudio histopatológico confirmó el diagnóstico de Tumor células de Leydig. Al cabo de un mes de la cirugía nuevos exámenes demuestran normalización de las concentraciones de todos los andrógenos que se mantiene después de 1 año de seguimiento (Tabla 1). Los Glucorticoides se retiraron en forma progresiva.
Tabla 1: Valores de andrógenos en distintas etapas del desarrollo del caso clínico.
Nota: 2014 debut, 2015-2016 desarrollo, 2017 (achurado) valores posteriores a cirugía.
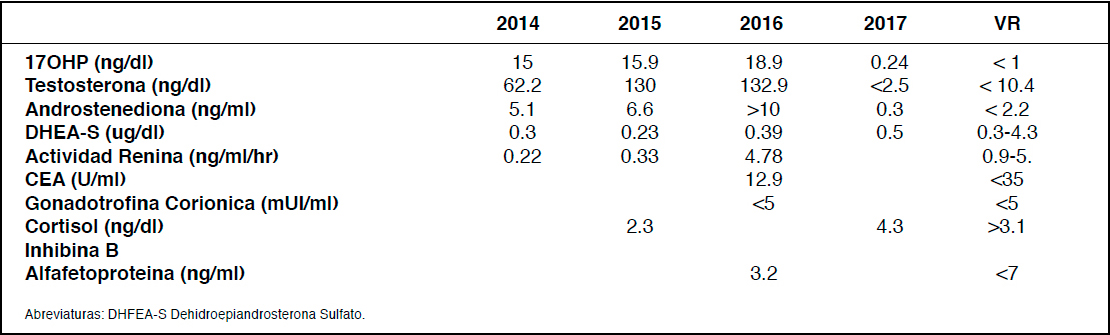
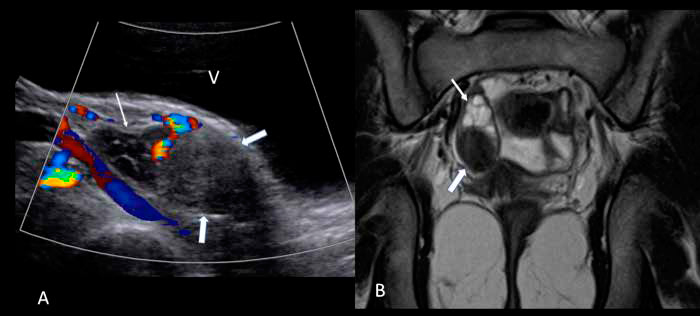 Figura 2. Determinación de los niveles séricos de A) Interleuquina-6 (IL-6); y B) proteína quimiotáctica de monocito-1 (MCP-1) en pacientes
con DMT1 y sobrepeso. Los datos representan el promedio ± SEM de la intensidad de fluorescencia media (IMF). Las diferencias entre el día
cero y después de 6 meses de adherencia al programa de cambios de estilo de vida se calcularon usando una prueba t de Student y Wilcoxon.
P <0.05 versus basal.
Figura 2. Determinación de los niveles séricos de A) Interleuquina-6 (IL-6); y B) proteína quimiotáctica de monocito-1 (MCP-1) en pacientes
con DMT1 y sobrepeso. Los datos representan el promedio ± SEM de la intensidad de fluorescencia media (IMF). Las diferencias entre el día
cero y después de 6 meses de adherencia al programa de cambios de estilo de vida se calcularon usando una prueba t de Student y Wilcoxon.
P <0.05 versus basal.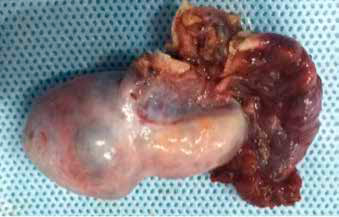 Figura 1. (A) Ultrasonografía pelviana muestra el ovario derecho aumentado de volumen, con una lesión sólida, heterogénea y sin flujo vascular en el polo inferior
(flechas gruesas) y folículos de aspecto normal en el polo superior (flechas delgadas). (B) RM de pelvis en corte coronal y en secuencia ponderada en T2, muestra
lesión hipointensa del ovario derecho (flechas gruesas) y los folículos en el polo superior (flechas delgadas).
Figura 1. (A) Ultrasonografía pelviana muestra el ovario derecho aumentado de volumen, con una lesión sólida, heterogénea y sin flujo vascular en el polo inferior
(flechas gruesas) y folículos de aspecto normal en el polo superior (flechas delgadas). (B) RM de pelvis en corte coronal y en secuencia ponderada en T2, muestra
lesión hipointensa del ovario derecho (flechas gruesas) y los folículos en el polo superior (flechas delgadas).Discusión
La PP es una variante del desarrollo puberal que en ocasiones puede corresponder a la primera manifestación de una patología suprarrenal u ovárica. Sí bien en la mayoría de los casos esta corresponde a activación fisiológica de la zona reticular de la glándula suprarrenal (adrenarquia) que se expresa antes del tiempo esperado, sin consecuencias negativas para la salud, la presencia de signos de virilización obliga a descartar otras causas como HSRNC, tumores (suprarrenal u ovárico) o contaminación con andrógenos externos.
Entre las causas patológicas la HSRCN por deficiencia de la enzima 21-Hidroxilasa por mutación del gen CYP21A2 es la etiología más común (90%), aunque existen otras mutaciones menos frecuentes como déficit de 3 beta Hidroxilasa, 11 Hidroxilasa y déficit de sulfatasa entre otras.
El diagnóstico de la HSRNC se sospecha por 17 OHP elevada y se confirma con estudio molecular. La 17 OHP se eleva por ser el andrógeno previo al bloqueo por deficiencia de la enzima. El hiperandrogenismo se explica por aumento de la hormona adrenocorticotrófica (ACTH) secundario a disminución del feedback producido por Cortisol, lo cual induce una mayor actividad de la maquinaria enzimática responsable de la esteroidogénesis suprarrenal, con consecuente aumento de los andrógenos, ya que las enzimas que intervienen en su síntesis no se encuentran bloqueadas.
La mayoría de niñas con HSRNC, no presentan virilización importante y la respuesta clínica y bioquímica al tratamiento con dosis fisiológicas de hidrocortisona es apropiada. Cuando no existe respuesta clínica ni bioquímica, y el estudio molecular es negativo para mutaciones del gen CY21A2, debe descartarse la presencia de un tumor productor de andrógenos, el que pueden ser de origen suprarrenal u ovárico.
Los tumores suprarrenales pueden también secretar cortisol y/o aldosterona; clínicamente se pueden asociar a Síndrome de Cushing, hipertensión arterial y virilización. Una elevación de DHEA-S orienta a que la fuente de los andrógenos es suprarrenal, pero nuestra paciente tenía concentraciones bajas de DHEA-S, asociado a concentraciones elevadas de T y A no suprimidas por el uso de corticoides; lo que hizo plantear un origen gonadal de los andrógenos en nuestro caso.
Las neoplasias ováricas (tanto benignas como malignas) dan cuenta de alrededor del 1% de los tumores en niños y adolescentes. La mayoría son de carácter benigno, sin embargo, la prevalencia de neoplasias malignas alcanza entre un 10- 30% en niñas y adolescentes1,3,4.
Los tumores ováricos se clasifican por la OMS según su histología y dependen de la edad de presentación. En mujeres entre 10 a 30 años son más frecuentes los tumores de células germinales, correspondiendo aproximadamente al 70% de los tumores ováricos en este grupo etario5; estos se presentan más temprano que los tumores epiteliales del ovario (que son el tipo más frecuente de tumor maligno) cuya edad promedio de presentación es de 50 años, y solo 12% lo hace antes de los 30 años6.
Los tumores ováricos de células Leydig pertenecen al grupo de los tumores del estroma gonadal y de los cordones sexuales, los cuales presentan un patrón de diferenciación testicular. Los pacientes que presentan tumores malignos del estroma y de los cordones sexuales en general se diagnostican en etapas tempranas como ocurrió en nuestro caso. Estos tumores son un grupo heterogéneo de neoplasias malignas y benignas poco frecuentes, corresponden a un 1,2% de los tumores ováricos primarios pero son los con mayores manifestaciones endocrinas, su incidencia anual en Estados Unidos es de 0,2/100.000 mujeres6,7. El diagnóstico se realiza mediante biopsia quirúrgica y la etapificación es la misma que el resto de los tumores ováricos primarios. El diagnóstico diferencial más importante además de la HSCNC son los tumores virilizantes de la glándula suprarrenal.
La Ecografía pelviana es el método de imágenes de elección cuando se sospecha una lesión anexial, con alta sensibilidad en la detección de una masa ovárica. En el caso de nuestra paciente, la lesión no se demostró en el examen inicial y sólo fue evidente en la Ecografía efectuada 2 años 9 meses más tarde, luego del hallazgo de la RM. La RM de abdomen es el método de elección ante la sospecha de una lesión suprarrenal.
Las células de Leydig participan en la síntesis de testosterona a través de la conversión de la molécula de colesterol. Dentro de los intermediarios previos a la testosterona se encuentra la 17 OHP que posteriormente es metabolizada a DHEA. La producción supra fisiológica de testosterona, de causa tumoral en este caso, requiere un aumento a de todos los precursores esteroidales de la síntesis de testosterona, esto explica que 17 OHP esté moderadamente elevada en nuestro caso, como está descrito en otros casos de tumores productores de andrógenos. Por otra parte la no disminución de andrógenos con el uso de análogo de LHRH, demuestra que estos a su vez son independiente del eje reproductivo.
Estudios genéticos recientes han reportado una relación entre mutaciones del gen DICER1, involucrado en el procesamiento de microRNAs, con la incidencia de tumores del estroma y de los cordones sexuales, así como también de otro tumor poco frecuente, el Blastoma Pleuropulmonar amiliar (OMIM 601200). Específicamente la mutación DICER1 RNase IIIb mutación “hotspot” se ha relacionado con la aparición de tumores. Además se ha descrito relación entre mutaciones de gen DICER1 y la presencia de nódulos tiroideos y cáncer de tiroides bien diferenciado (OMIM 138800)8. Las mutaciones de este gen tienen una prevalencia de entre un 50%-60% en los tumores de Sertoli-Leydig8.
No se ha demostrado relación entre la presencia de esta mutación y el grado de malignización o recurrencia9. Los tumores del estroma y de los cordones sexuales son tumores de baja incidencia por lo que su historia natural solo se debe levantar a partir de estudios multicéntricos.
Hasta nuestro conocimiento, la evidencia disponible de tumores de Leydig aislados en niños es muy escasa, existiendo mayor cantidad de publicaciones sobre tumores de Sertoli-Leydig (Tumor mixto de ambos tipos celulares). Si bien este no es exactamente el mismo tipo celular de nuestro caso clínico, se puede especular que tratándose de un tumor mixto con componentes de células de Leydig, que pertenece a la categoría de tumores provenientes del estroma y de los cordones sexuales, presente elementos en común con los tumores puros de células de Leydig.
El pronóstico a largo plazo de estos tumores es, en general bueno, teniendo una sobrevida a 5 años de entre un 70-90%10. El principal indicador pronóstico de los tumores de Leydig y Sertoli es el grado de diferenciación histológica y su etapificación11.
Conclusiones
Este caso clínico ilustra como un tumor ovárico puede ser de difícil diagnóstico clínico, bioquímico e imagenológico. Lo más importante en nuestra paciente fue el seguimiento, que permitió apreciar que la virilización y las elevadas concentraciones de testosteronas no se frenaron con el uso de corticoides (lo que descarta su dependencia de ACTH), como tampoco con el uso de análogos de LHRH (descartá su dependencia de gonadotrofinas). Este comportamiento permitió insistir en la búsqueda de un tumor, el cual logró confirmarse recién 2 años 9 meses después de haber iniciado su cuadro clínico. También nos enseña que frente a una niña con PP y signos de virilización progresiva a pesar de tratamiento corticoidal, se debe insistir en la búsqueda de un tumor suprarrenal o gonadal, especialmente si el estudio genético ha descartado las formas más frecuentes de hiperplasia suprarrenal. En estos casos una elevación moderada de 17 OHP, es de carácter inespecífico.
Referencias
- Oltmann SC, Garcia N, Barber R, Huang R, Hicks B, Fischer A. Can we preoperatively risk stratify ovarian masses for malignancy? J Pediatr Surg 2010; 45(1): 130-134.
- Cass DL, Hawkins E, Brandt ML, Chintagumpala M, Bloss RS, Milewicz AL, et al. Surgery for ovarian masses in infants, children, and adolescents: 102 consecutive patients treated in a 15-year period. J Pediatr Surg 2001; 36(5): 693-699.
- Brown MF, Hebra A, McGeehin K, Ross AJ 3rd. Ovarian masses in children: a review of 91 cases of malignant and benign masses. J Pediatr Surg 1993; 28(7): 930-933.
- Hermans AJ, Kluivers KB, Wijnen MH, Bulten J, Massuger LF, Coppus SF. Diagnosis and treatment of adnexal masses in children and adolescents. Obstet Gynecol 2015; 125(3): 611-615.
- Zalel Y, Piura B, Elchalal U, Czernobilsky B, Antebi S, Dgani R. Diagnosis and management of malignant germ cell ovarian tumors in young females. Int J Gynaecol Obstet 1996; 55(1): 11-10.
- Quirk JT, Natarajan N. Ovarian cancer incidence in the United States, 1992-1999. Gynecol Oncol 2005; 97(2): 519-523.
- Colombo N1, Peiretti M, Garbi A, Carinelli S, Marini C, Sessa C, et al. Non-epithelial ovarian cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012; 23: vii20-vii26.
- Heravi-Moussavi A, Anglesio MS, Cheng SW, Senz J, Yang W, Prentice L, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med 2012; 366(3): 234-242.
- Young RH. Sertoli-Leydig cell tumors of the ovary: Review with emphasis on historicalaspects and unusual variants. Int J Gynecol Pathol 1993; 12(2): 141-147.
- Litta P, Saccardi C, Conte L, Codroma A, Angioni S, Mioni R. Sertoli- Leydig cell tumors: Current status of surgical management: Literature review and proposal of treatment. Gynecol Endocrinol 2013; 29(5): 412- 417.
- Schneider DT, Orbach D, Cecchetto G, Stachowicz-Stencel T, Brummel B, et al. Ovarian Sertoli Leydig cell tumours in children and adolescents: An analysis of the European Cooperative Study Group on Pediatric RareTumors (EXPeRT). Eur J Cancer 2015; 51(4): 543-550.