Síndrome de insensibilidad a andrógenos: Revisión bibliográfica a propósito de 5 casos
José F. Delgado G.1,2 https://orcid.org/0000-0002-1865-7196
Javiera Pérez E.1,2 https://orcid.org/0000-0001-5410-2860
Dasha Delgado B.3 https://orcid.org/0000-0001-6549-6471
Felipe Valenzuela P.1 https://orcid.org/0000-0002-3631-5704
Alejandra Martínez G.1 https://orcid.org/0000-0002-8484-8126
Eugenio Arteaga U.1* https://orcid.org/0000-0002-8090-539X
Androgen insensitivity syndrome: literature review about 5 cases
1. Médico, Departamento de Endocrinología y CETREN-UC, Escuela de Medicina, Pontificia Universidad Católica de Chile, Santiago, Chile.
2. Médico, Servicio de Endocrinología Hospital Dr. Sótero del Río y Hospital Dipreca, Santiago, Chile.
3. Ayudante alumna, Hospital Dr. Sótero del Rio, Santiago, Chile.
*Correspondencia: Eugenio Arteaga / eugenioarteaga@gmail.com
Departamento de Endocrinología, Escuela de Medicina, Pontificia Universidad Católica de Chile
Diagonal Paraguay 362, Santiago Centro, Chile.
Trabajo sin financiamiento
Recibido: 21-07-2020
Aceptado: 08-10-2020
Resumen: El síndrome de insensibilidad a andrógenos (AIS en la sigla inglesa) es una entidad muy poco frecuente en endocrinología. Se caracteriza por la mutación del receptor de andrógenos de magnitud variable, por medio del cual individuos 46,XY no se virilizan normalmente, a pesar de conservar sus testículos y tener concentraciones de testosterona en rango masculino. El cuadro clínico es variable y depende la profundidad de la alteración del receptor. En un extremo, hay casos de insensibilidad androgénica completa (CAIS) con fenotipo femenino. En el otro extremo hay insensibilidad parcial (PAIS) que se extiende desde el fenotipo femenino, con o sin ambigüedad genital, hasta los casos de hombres infértiles o con subvirilización, que presentan insensibilidad androgénica más leve. En los fenotipos femeninos, los testículos suelen estar en posición ectópica y aquellos ubicados dentro del abdomen tienen riesgo de malignizarse, por lo que suelen extirparse. Estos son los casos de más difícil manejo, pues aparte de la necesidad de gonadectomía seguida de terapia hormonal femenina, existe una vagina estrecha y en fondo de saco ciego y que suele requerir corrección quirúrgica para permitir la actividad sexual. En este trabajo presentamos 5 casos de AIS vistos recientemente en 2 centros clínicos de Santiago y que ilustran la heterogeneidad de presentación. Además, hacemos una revisión actualizada de los criterios diagnósticos, los tratamientos más adecuados y el manejo global de esta condición.
Palabras clave: Amenorrea primaria; Desorden desarrollo sexual; Insensibilidad a andrógenos; Receptor de andrógenos; Testículos.
Abstract: The Androgen insensitivity syndrome (AIS, in its English acronym) is a very rare entity in endocrinology. It is characterized by a variable magnitude androgen receptor mutation, whereby 46, XY individuals are not normally virilized, despite retaining their testicles and having testosterone concentrations in the male range. The clinical picture is variable and depends on the depth of the receptor alteration. At one extreme, there are cases of complete androgenic insensitivity (CAIS) with a female phenotype. At the other extreme, there is partial insensitivity (PAIS) that extends from the female phenotype, with or without genital ambiguity, to cases of infertile or undervirilized men, who have milder androgenic insensitivity. In female phenotypes, the testes are usually in an ectopic position and those located within the abdomen are at risk of malignancy, and therefore are usually removed. These are the most difficult cases to manage because apart from the need for gonadectomy followed by female hormonal therapy, there is a narrow vagina and a deep blind pouch that usually requires surgical correction to allow sexual activity. In this work, we present 5 cases of AIS recently seen in 2 clinical centers in Santiago and that illustrate the heterogeneity of presentation. In addition, we make an updated review of the diagnostic criteria, the most appropriate treatments, and the overall management of this condition.
Key words: Androgen insensitivity; Androgen receptor; Disorder of sexual development; Primary amenorrhea; Testicles.
Los defectos en la función del receptor de andrógenos (RA) causan un desorden del desarrollo sexual (DSD en la sigla inglesa) en el cual individuos XY no se virilizan según el patrón normal, a pesar de la presencia de testículos y concentraciones de testosterona en rango masculino1. Este cuadro se ha denominado síndrome de Insensibilidad a Andrógenos (AIS), en reemplazo de la antigua denominación de Testículo Feminizante, que era inexacta y mal interpretable.
Los AIS presentan un continuo clínico que va desde la ausencia total hasta la disminución leve de los efectos androgénicos, y cuya presentación clínica se extiende desde un fenotipo femenino hasta un fenotipo masculino con sub virilización o infertilidad, respectivamente.
Nuestro objetivo es presentar 5 casos de AIS vistos en los últimos años en 2 centros clínicos de Santiago, y hacer una revisión sobre esta enfermedad.
Casos Clínicos
Caso 1
Niña de 16 años que consulta por amenorrea primaria y con un fenotipo femenino con mamas Tanner 4 pero ausencia de vello axilar y pubiano. No tenía antecedentes familiares ni personales relevantes. Sus exámenes demuestran cariotipo 46,XY, FSH 3.1 mUI/mL (valor normal masculino [VNM] 1,5- 12,4) Testosterona 460 ng/dL (VMN 180-763) Estradiol 25.8 pg/mL (VNM 25.8-60,7), SHBG 17,5 nmol/L (VNM 16.5-55,9).
La RM de abdomen y pelvis (Figura 1) demuestra agenesia uterina y ausencia de gran parte de cavidad vaginal, reconociendo 2 nódulos de aspecto solido homogéneos en excavación pélvica (posición anexial) con señal sugerente de testículos, con diámetro mayor de 31 mm a derecha y de 21 mm a izquierda.
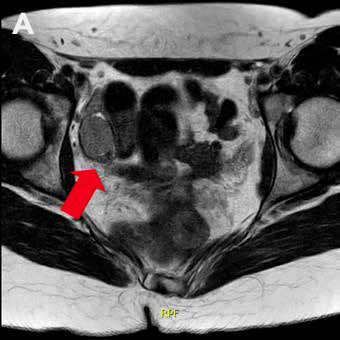
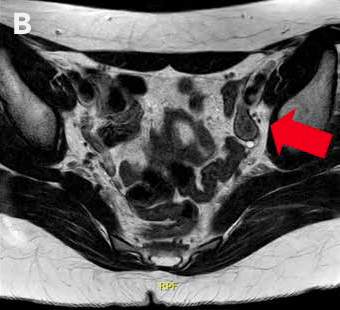
Figura 1: RM de pelvis que muestra 2 testículos intra-abdominales, A. Testículo derecho, B. Testículo izquierdo.
Caso 2
Mujer de 27 años, con antecedente de amenorrea primaria con hábito femenino, mamas Tanner V, escaso vello no terminal axilar y púbico, y que tenía pareja heterosexual y vida sexual normal. Sus exámenes hormonales demostraron testosterona en rango masculino (Tabla 1) y cariograma 46,XY compatible con insensibilidad a andrógenos. La TAC de pelvis mostró ausencia de útero, una imagen ovoidea con densidad de partes blandas de 27 mm de diámetro mayor a nivel de la bifurcación de los vasos iliacos derechos interpretada como ovario normal, y en situación anexial izquierda una masa sólida de 90 mm de diámetro mayor, bien delimitada algo heterogénea sin calcificaciones, sin liquido libre ni adenopatías (Figura 2). Fue sometida a gonadectomía bilateral 4 años antes de la consulta y la biopsia demostró un seminoma espermatocítico de 11 cm asociado a un foco de neoplasia germinal intratubular de 2 mm en la gónada izquierda y un testículo atrófico y fibroso, con epidídimo de aspecto conservado a derecha.
Tabla 1. Exámenes hormonales paciente 2.
| VN hombres | Pre- cirugía | Post-cirugía | |
| FSH mUI/mL | (1,5-9,7) | 36,6 | 79,9 |
| LH mUI/mL | (1,3-10,5) | 31,6 | 62,8 |
| Testosterona ng/dL | (132-813) | 731 | 20,40 |
| Alfa fetoproteínas (ng/ml) | (< 7,2) | 1,51 | |
| βhCG (mUI/ml) | (<4,8) | <2,39 |
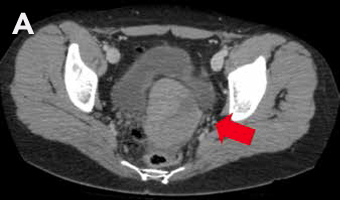
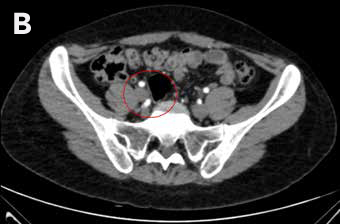
Figura 2: TAC de pelvis que muestra una gónada derecha y una masa anexial izquierda, A Masa anexial, B. Gónada derecha.
Caso 3
Mujer de 19 años que consulta para corrección de su estrechez vaginal. Tiene antecedente que, a los 12 años, sin haber iniciado desarrollo puberal, se le efectúa una cirugía de urgencia por sospecha de hernias inguinales bilaterales atascadas, extirpándose sendas gónadas cuyo estudio histopatológico demuestra testículos con retardo del desarrollo bilateral. No hubo estudio hormonal previo a ello pero el cariograma fue 46,XY, compatible con insensibilidad a andrógenos.
Sus antecedentes personales eran irrelevantes, pero en su familia había 2 tías y dos primas de la línea materna con insensibilidad a andrógenos. El estudio hormonal post cirugía demostró un hipogonadismo hipergonadotropo (FSH: 110.6 mUI/ml. LH: 43.29 mUI/ml, E2: 11.28 pg/ml).
Se inició estrogenoterapia en dosis ascendente de estradiol oral hasta alcanzar una dosis de 2 mg diario. Con ello hubo crecimiento pondoestatural hasta 166 cm a los 17 años, superando su expectativa de talla genética. Las mamas se desarrollaron desde Tanner I a IV, pero hipoplásicas; nunca hubo desarrollo de pelo púbico. Sus genitales externos eran de aspecto femenino. Había ausencia completa de introito vaginal, se reconocía clítoris de aspecto normal y labios menores, meato uretral muy pequeño y piel que se fusionaba entre labios mayores y periné, extendiéndose hacia el ano.
La paciente fue derivada a atención sicológica y a un centro de referencia para corrección de su acortamiento vaginal por su deseo de iniciar actividad sexual.
Caso 4
Mujer de 53 años que consulta para manejo de su terapia hormonal de la menopausia. Ella no conserva sus exámenes de 30 años atrás, pero relata que consultó por amenorrea primaria a los 14 años. El examen físico y radiológico habría demostrado una vagina ciega, vello púbico escaso, ausencia de útero y clitorimegalia. El laboratorio demostró testosterona en rango masculino y el cariograma fue 46,XY.
Con el diagnóstico de Insensibilidad androgénica parcial, o PAIS en la sigla inglesa, se le sometió a la extirpación de 2 testículos intra-abdominales y escisión del clítoris hipertrofiado y se le inició desde entonces terapia estrogénica. Además, se le colocaron prótesis mamarias bilaterales. Nunca se corrigió su acortamiento vaginal lo que le impidió tener actividad sexual. Actualmente tiene pareja homosexual.
En el examen físico actual se observa hábito femenino, mamas protésicas, vello axilar ralo y vello púbico Tanner III. El examen ginecológico demuestra la escisión del clítoris, genitales externos intactos y la vagina es muy estrecha, permitiendo solo la introducción del dedo índice, hasta un máximo de 3 cm. de profundidad.
Caso 5
Mujer de 32 años que consulta por amenorrea primaria. Relataba que en controles previos se le había demostrado ausencia de genitales internos femeninos confirmado por ecotomografía. Al nacer las gónadas se palpaban en la región vulvar y había genitales ambiguos (tubérculo genital de 1,8 cm con meato urinario hipospádico y labios escrotales hiperpigmentados fusionados en su porción posterior, pequeña apertura vaginal, clasificado como estadio III-IV de Prader). El cariotipo fue 46,XY. A los 9 meses se realizó gonadectomía bilateral, y a los 7 años plastia de labios mayores y clítoris. A los 15 años inició terapia de inducción puberal y se ha mantenido en reemplazo hormonal estrogénico con adecuado desarrollo de caracteres sexuales secundarios. A los 18 años se realizó cirugía de construcción de neovagina por vía laparoscópica.
El examen físico actual demuestra fenotipo femenino con mamas Tanner IV, sin vello axilar ni púbico. En la tabla 2 se muestra estudio hormonal realizado a los 14 días de vida.
Se presentan en la tabla 3 el fenotipo, las características clínicas, valores de testosterona y diagnóstico de los 5 casos clínicos.
Tabla 2. E xámenes de laboratorio de paciente 5, a los 14 días de vida.
| VN hombres 0-12 meses | Paciente | |
| Estradiol pg/mL | < 24 | 12 |
| LH mUI/mL | 0,16-4,1 | 0,9 |
| FSH mUI/mL | 0,5-4,5 | 1,5 |
| Testosterona ng/dL | 75-400 | 190 |
| Cortisol ug/dL | > 15 | 17 |
| 17-OH progesterona ng/mL | < 2,0 | 0,7 |
| AMH ng/mL | 24-275 | 130 |
Tabla 3. Resumen de los 5 casos.
| Caso 1 | Caso 2 | Caso 3 | Caso 4 | Caso 5 | |
| Fenotipo | Femenino | Femenino | Femenino | Femenino | Femenino |
| Motivo de consulta | Amenorrea primaria | Dolor y tumor pélvico | Dolor inguinal bilateral | Amenorrea primaria | Genitales ambiguos |
| Mamas | Tanner IV | Desarrollo normal | Hipoplasia | Hipoplasia | Tanner IV por Inducción puberal |
| Vello sexual | Ausente | Escaso | Ausente | Escaso | Ausente |
| Genitales externos | Femenino, vagina corta | Femenino, sin disfunción sexual | Femenino con ambigüedad | Femenino, clitorimegalia y vagina corta | |
| Gónadas | Pelvis | Pelvis | Inguinal | Pelvis | Región vulvar |
| Testosterona (ng/dL) | 460 | 731 | Rango | Rango masculino | 190 |
| Diagnóstico IAS | CAIS | CAIS | PAIS | PAIS | PAIS |
Discusión
El objetivo de este trabajo es presentar 5 casos con insensibilidad a andrógenos vistos recientemente en dos centros de salud de la región metropolitana, destacando la heterogeneidad de las formas de presentación y hacer una revisión del estado del arte respecto a esta patología.
Patogénisis
El síndrome de resistencia a andrógenos (AIS en su sigla inglesa) se debe a una mutación del gen que codifica para el receptor de andrógenos (RA), localizado en el cromosoma Xq11-12. Esta mutación hace que el RA pierda su función total o parcialmente. Se presenta en individuos 46,XY que tienen testículos funcionantes, descendidos o no y que secretan testosterona. La presencia de AMH determina inhibición de las estructuras Mullerianas (útero, trompas y parte superior de la vagina) y la deficiente acción de testosterona permite que se desarrolle una mujer fenotípica en los casos más intensos. El espectro clínico depende de la magnitud de la pérdida del efecto de testosterona, variando de un fenotipo femenino con vello axilar y pubiano ausente o muy escaso, en un extremo, a un fenotipo masculino con subvirilización e infertilidad, en el otro. A pesar de la variabilidad de expresión fenotípica, los pacientes tienen características endocrinas, genéticas y fisiopatología semejantes2.
Presentación clínica
Dependiendo de la magnitud de la pérdida de acción androgénica se describen formas completas e incompletas de insensibilidad a andrógenos (Tabla 4):
1. Insensibilidad completa a andrógenos (CAIS)
Es la tercera causa en frecuencia de amenorrea primaria, después de las disgenesias gonadales y la ausencia de vagina. Su incidencia puede llegar a 1: 20.000-1: 99.0003. Suele diagnosticarse en niñas adolescentes con amenorrea primaria, fenotipo absolutamente femenino, con desarrollo mamario completo, a excepción de vello axilar y pubiano ausente o muy escaso. Respecto a la estatura final, esta se rige más por un patrón masculino. La identificación de género es femenina en la mayoría de los casos, aunque hay excepciones. Dado que tienen vaginas cortas, suele ser necesaria la cirugía para elongarla y permitir la actividad sexual4.
También puede diagnosticarse al nacimiento o en la infancia por la presencia de masas inguinales (que contienen testículos) o hernias en niñas aparentemente normales, con genitales externos normales.
Los testículos suelen estar ubicados en la pelvis (como nuestros casos 1, 2 y 4), canales inguinales (caso 3) o en los labios mayores (caso 5). La histología muestra lo mismo que los testículos no descendidos, es decir células de Leydig normales o aumentadas, pero sin espermatogénesis. Prácticamente no existen estructuras de Müller, aunque en ocasiones puede haber remanentes uterinos5.
El patrón hormonal puede no diferenciarse de lo normal en la infancia, pero en la adolescencia suele encontrarse testosterona en rango masculino normal o alto en comparación con varones post puberales y la LH elevada demostrando la insensibilidad hipotálamo-hipofisaria a la retroalimentación negativa de testosterona6.
2. Insensibilidad parcial a andrógenos (PAIS)
La incidencia es de 1: 130.000, haciendo necesaria la secuenciación del gen para un diagnóstico correcto. Considera casos en que el defecto de la acción de andrógenos es menos grave y da origen a una variedad de desórdenes del desarrollo sexual 46,XY que van desde mujeres con grados moderados de virilización hasta el caso de hombres fértiles pero sub virilizados o con ginecomastia7.
Existen varias formas de presentación del PAIS
- Fenotipo femenino con virilización leve. Las mujeres en este subgrupo se parecen a las mujeres con CAIS, pero tienen un patrón de pelo secundario femenino, aunque puede ser ralo, y puede haber virilización parcial de los genitales en la pubertad8. Además, puede haber fusión parcial de los pliegues labio escrotales, como en nuestro caso 3, o clitorimegalia como el caso 4, que fue resecado quirúrgicamente por un evidente error del juicio médico. Puede haber algún desarrollo de ductos de Wolff como epidídimos, vasa deferente, vesículas seminales, ductos eyaculatorios y próstata.
- Fenotipo masculino predominante. Son niños 46,XY que tienen defectos variables en su virilización9. Puede haber hipospadias peri escrotales y escroto bífido con testículos in situ. Sus genitales externos pueden variar desde micropene con uretra normal hasta una fusión escrotal incompleta y seudovagina. En la pubertad suele aparecer ginecomastia marcada y persistente y que suele requerir cirugía y en la vida adulta son infértiles. Su pelo púbico, axilar y facial es ralo. Estos varones no suelen tener conflictos en la identidad de género y pueden desarrollar actividad sexual satisfactoria.
- Síndrome de hombre infértil. (también clasificado como MAIS, del inglés mild). Solo presentan infertilidad, leve sub-virilización y ginecomastia. De hecho, muchos hombres con azoospermia u oligospermia severa han demostrado defectos en el receptor de andrógenos. Su hábito corporal y desarrollo de pelos es absolutamente masculino y sus testículos están en los escrotos.
- Síndrome de hombre fértil sub virilizado (MAIS, del inglés mild). Es muy poco frecuente y se caracteriza por hombres fértiles, pero algo sub virilizados (menor pelo facial y corporal, ginecomastia y eventualmente micropene con testículos descendidos y con recuento espermático presente). Su orientación sexual es masculina.
- Atrofia muscular espino bulbar (también es clasificado como MAIS). También se le denomina enfermedad de Kennedy y es una variante casi desconocida de PAIS10. Se caracteriza por degeneración progresiva de las neuronas motoras anteriores, ginecomastia tardía, espermatogénesis defectuosa y patrón hormonal compatible con resistencia androgénica.
La incidencia y prevalencia de las formas menos frecuentes de AIS como síndrome de hombre infértil u hombre fértil sub-virilizado es desconocida pues muchos casos son clínicamente indetectables, a menos que se sometan a estudio molecular del RA, lo que no está disponible en la mayoría de los centros clínicos.
Tabla 4. Características fundamentales de los distintos tipos de síndrome de Insensibilidad a andrógenos.
| Características Clínicas | Insensibilidad andrógenos completa (CAIS) | Mujer/insensibilidad parcial (PAIS) | Hombre/Insensibilidad parcial (PAIS) | Insensibilidad andrógenos leve (MAIS) |
| Genitales externos | Completamente femenina, vagina corta | Clitorimegalia, vagina estrecha, fusión variable labios menores | Hipospadias, escroto bífido | Completamente masculinos |
| Localización gónadas | Abdominal o inguinal | Inguinal | Escrotal | Escrotal |
| Desarrollo puberal | Feminizante | Desarrollo mamario con leve virilización | Ginecomastia persistente, leve virilización | Ginecomastia y virilización parcial o total |
| Vello sexual | Escaso o ausente | Escaso | Ligeramente disminuido | Ligeramente disminuido |
| Espermatogénesis | Ausente | Ausente | Ausente | Gravemente afectada |
Genética
La mayor parte de la información viene de pacientes con CAIS. Las mutaciones del receptor de andrógenos incluyen deleciones, anormalidades de splicing, codón de término prematuro y mutaciones sin sentido11.
Esta última es la más frecuente y alcanza al 80% de los casos, determinando sustitución de aminoácidos. La deleción completa del gen AR solo se reporta en el 1%. Alrededor del 40% de los casos de CAIS no tienen antecedentes familiares, lo que no obsta para que en sus antepasados haya portadores de la mutación expresada en el caso índice. En el resto puede haber antecedente familiar, como fue muy destacado en el caso 3.
La correlación genotipo-fenotipo es pobre. Las deleciones y los codones de término prematuro solo se han visto en CAIS. Pero las sustituciones de aminoácidos, que son las alteraciones más frecuentes, pueden causar todos los fenotipos, desde hombres infértiles a mujeres con CAIS.
Esta pobre correlación genotipo-fenotipo se ejemplifica en casos en una misma familia que presentando la misma mutación tienen distinto fenotipo12.
Sospecha clínica de AIS
Los siguientes cuadros clínicos deben hacer sospechar un síndrome de insensibilidad de andrógenos
- Hombres y mujeres de todas las edades con genitales atípicos.
- Mujeres con hernias inguinales o masas en labios mayores.
- Mujeres con amenorrea primaria, desarrollo puberal normal, pero vello sexual ausente o disminuido.
- Mujeres adolescentes con virilización y clitorimegalia.
- Hombres adolescentes que no completan la pubertad normal o tienen ginecomastia persistente o signos de sub-virilización.
- Hombres adultos sub virilizados o infértiles asociados a azoospermia o oligospermia severa.
En relación con nuestros casos, nuestras pacientes cumplían con los criterios 1 a 4 de e stos cuadros.
Alteraciones Hormonales
El patrón hormonal es similar en todas las formas de AIS aunque ha sido más estudiada en los casos de CAIS. La testosterona sérica está en el rango de niños u hombres normales de edad equivalente, o incluso más elevada. La LH suele estar elevada como expresión de la resistencia de LH al feedback negativo de los andrógenos a nivel hipotálamo-hipofisario. Ello determina un estímulo exagerado sobre la síntesis testicular de testosterona. La FSH igual que la inhibina B suele estar dentro de rango normal13, aunque puede haber excepciones, tal como ocurrió en nuestra paciente 2 que tenía FSH elevada al diagnóstico, posiblemente por atrofia gonadal ectópica.
Los niveles de estrógenos suelen estar dentro de rango normal masculino, independiente del patrón de feminización de estas mujeres. La ginecomastia en los sujetos con fenotipo masculino de PAIS debiera por tanto atribuirse a un desbalance entre los valores normales de estrógenos para un varón y la acción androgénica disminuida.
En las mujeres con PAIS con virilización leve y en los hombres con PAIS el patrón hormonal es semejante al de las mujeres con CAIS; en el subtipo PAIS de hombre infértil, el patrón hormonal es menos alterado.
En pacientes con CAIS o PAIS, la hormona antimülleriana (AMH) o la inhibina B son normales o sobre lo normal para el rango masculino, confirmando la presencia de testículos funcionales13.
Diagnóstico diferencial de pacientes AIS
En las formas completas es más fácil el diagnóstico, pero en formas parciales el diagnóstico diferencial puede ser más complejo y deben considerarse defectos globales en la función testicular (disgenesias gonadales) y los defectos en la síntesis de andrógenos:
- Disgenesia gonadal mixta: está asociada con cariotipo mosaico 45,X/46,XY, presenta testes disgenéticos en un lado y estría gonadal en el otro; la presencia de una gónada descendida eleva la sospecha de esta condición. Puede haber remanentes müllerianos con niveles bajos de testosterona y AMH.
- Disgenesia gonadal completa o parcial: en la forma completa son mujeres con presencia de estructuras müllerianas, estrías gonadales que explican los niveles bajos de testosterona, AMH inhibina B; mientras que en las formas parciales el fenotipo va desde infertilidad, hipospadias, genitales ambiguos a hombres subvirilizados.
- Defectos en la síntesis de andrógenos, que se caracterizan por niveles bajos de testosterona o dihidrotestosterona (DHT) a pesar de estimulación adecuada por LH que suele estar elevada, lo que puede ocurrir por defectos en el receptor de LH, por defectos en algunas de las enzimas de la vía de síntesis de colesterol a testosterona, o por deficiencia de la 5-alfa reductasa que afecta la conversión de Testosterona a DHT. Tienen fenotipo femenino o de hombres sub-virilizados. Estos casos son de transmisión autosómica recesiva.
Tratamiento
Lo primero a considerar es la adecuada asignación de sexo en el recién nacido. En ello es importante el fenotipo al nacer, la ambigüedad sexual, el diagnóstico genético, el pronóstico respecto al desarrollo puberal futuro, el riesgo de malignidad y la fertilidad. Es importante destacar que la presencia de un cariograma 46,XY, no es sinónimo de sexo masculino y se debe compartir con la familia y con el/la paciente la toma de decisiones, por lo que se recomienda diferir toda decisión médica o quirúrgica no urgente hasta que el niño/niña sea capaz de participar en un proceso compartido de toma de decisiones.
En los casos CAIS el fenotipo es femenino y es adecuada la asignación de dicho sexo. Además, muchas de estas niñas acceden al diagnóstico etiológico al llegar a la pubertad y consultar por amenorrea primaria, habiendo vivido hasta entonces como niñas y suelen tener orientación sexual femenina. En los casos PAIS, la presencia de ambigüedad sexual puede permitir un diagnóstico más precoz, pero si tienen fenotipo femenino y han sido criadas como niñas, el concepto anterior se aplica también a esta variedad.
Existe consenso en la literatura en recomendar que los pacientes CAIS se permita completar el desarrollo puberal previo a la gonadectomía por el riesgo de malignidad de 2-5% en mayores de 25 años14, mientras que en las mujeres PAIS el riesgo de malignidad puede ser mayor del 38%15, por lo que en estos casos se ha recomendado la gonadectomía antes de la fecha esperada del inicio puberal, evitando además la virilización parcial durante la pubertad.
Sin embargo, hay autores que proponen solo la extirpación en casos seleccionados16.
El riesgo de malignidad tiene reportes muy variables, sin embargo, cuando se consideran solo los casos AIS confirmados por estudio genético el riesgo de neoplasia in situ de células germinales es 10-15% y solo una minoría progresan a tumores testiculares invasores17.
Hoy se plantea un proceso de toma de decisión compartida relacionada a la preservación o extirpación gonadal, después de descartar las disgenesias gonadales que tienen mayor riesgo de malignidad18.
Para las pacientes que deciden preservar las gónadas, se recomienda RM o ultrasonido basal y luego anual mientras las gónadas parezcan normales; en presencia de masas, quistes, calcificaciones, adenopatías de etiología no explicables, cambios asimétricos del tamaño gonadal, se debe planear la biopsia laparoscópica o su extirpación. En nuestros casos se decidió por la gonadectomía post puberal en 3 y la extirpación más precoz en 1 debido a la sospecha de hernias inguinales complicadas. La paciente 1 se encuentra en seguimiento y no se ha decidido su gonadectomía.
Respecto a la cirugía urogenital, en mujeres se recomienda el uso de dilatadores o la vaginoplastía antes del inicio de la vida sexual; mientras que en los hombres las hipospadias deben ser corregidas lo antes posible luego del diagnóstico.
La ginecomastia de los varones con PAIS no es reversible, a diferencia de la ginecomastia puberal fisiológica, por ello se recomienda su extirpación precoz, especialmente cuando altera la percepción corporal.
Respecto a la terapia hormonal, después de la gonadectomía estas pacientes alcanzan un estado de hipogonadismo hipergonadotropo a edad muy temprana.
Por ello recomendamos que se extrapolen todos los conceptos del tratamiento de mujeres con falla ovárica prematura, pero sin utilización de progestinas dada que carecen de útero. Se recomienda utilizar dosis ascendentes de estradiol oral desde 0.5 mg /día hasta alcanzar en 2 años una dosis de mantención de 2 mg/día o e incluso más en casos seleccionados dependiendo de la edad y síntomas, considerando manejo individualizado. Aunque hay menor experiencia, la vía transdérmica parece una opción más fisiológica dado que evita la primera pasada hepática propia de la vía oral. En hombres con PAIS en edad adolescente se debe utilizar altas dosis de testosterona para permitir crecimiento peneano y mejorar otros signos de virilización.
Algunos estudios muestran que un 85% de las mujeres con CAIS cumplen criterios para trastornos psiquiátricos, 54% para trastornos ansiosos y 64% ha buscado ayuda psiquiátrica en el pasado19. Las familias de estos pacientes deben ser educadas inmediatamente luego de realizar el diagnóstico, informando a los pacientes de manera gradual pero completa acerca de su diagnóstico según la edad, con el fin de evitar el trauma temprano que puede implicar el conocer tempranamente la plenitud del diagnóstico. En nuestro caso solemos informar precozmente de la ausencia de útero con la consecuente infertilidad y en los casos con fenotipo femenino reforzamos el concepto de que son mujeres, independiente de cariotipo e histología de sus gónadas.
En conclusión, presentamos 5 casos de síndrome de insensibilidad a andrógenos que fueron diagnosticados por amenorrea primaria con desarrollo puberal incompleto en 3 casos, en una niña por masas inguinales que correspondieron a testículos y en 1 caso por ambigüedad genital y gónadas en región vulvar. Estos casos muestran la heterogeneidad de presentación de los cuadros de AIS. A raíz de esta casuística, presentamos una revisión actualizada de este trastorno genético/hormonal y proponemos las estrategias terapéuticas más apropiadas.
Referencias
- Hughes IA, Houk C, Ahmed SF, Lee PA. Consensus statement on management of intersex disorders. Consensus Group, ESPE Consensus Group Arch Dis Child. 2006; 91(7): 554.
- Quigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, French FS. Androgen receptor defects: historical, clinical, and molecular perspectives. Endocr Rev. 1995; 16(3): 271.
- Boehmer AL, Brinkmann O, Bruggenwirth H, et al. Genotype versus phenotype in families with androgen insensitivity syndrome. J Clin Endocrinol Metab 2001; 86:4151
- Wisniewski AB, Migeon CJ, Meyer-Bahlburg HF, Gearhart JP, Berkovitz GD, Brown TR, Money J. Complete androgen insensitivity syndrome: long-term medical, surgical, and psychosexual outcome. J Clin Endocrinol Metab. 2000; 85(8): 2664.
- Dodge ST, Finkelston MS, Miyazawa K. Testicular feminization with incomplete Müllerian regression. Fertil Steril. 1985; 43(6): 937.
- Doehnert U, Bertelloni S, Werner R, Dati E, Hiort O. Characteristic features of reproductive hormone profiles in late adolescent and adult females with complete androgen insensitivity syndrome. Sex Dev. 2015; 9(2): 69-74.
- Chu J, Zhang R, Zhao Z, Zou W, Han Y, Qi Q, Zhang H, Wang JC, Tao S, Liu X, Luo Z. Male fertility is compatible with an Arg(840)Cys substitution in the AR in a large Chinese family affected with divergent phenotypes of AR insensitivity syndrome. J Clin Endocrinol Metab. 2002; 87(1): 347.
- Morris JM, Mahesh VB. Further observations on the syndrome “Testicular feminization” Am J Obstet Gynecol. 1963; 87: 731.
- Wilson JD, Harrod MJ, Goldstein JL, Hemsell DL, MacDonald PC. Familial incomplete male pseudohermaphroditism, type 1. Evidence for androgen resistance and variable clinical manifestations in a family with the Reifenstein syndrome. N Engl J Med. 1974; 290(20): 1097.
- Pinsky L, Beitel LK, Trifiro MA. Spinobulbar muscular atrophy. En: The Metabolic and Molecular Bases of Inherited Disease, 8th, Scriver CR, Beaudet AL, Sly WS, Valle D (Eds), McGraw-Hill, New York 2001; 4147.
- Audí L, Ahmed SF, Krone N, Cools M, McElreavey K, Holterhus PM, Greenfield A, Bashamboo A, Hiort O, Wudy SA, McGowan R. Genetics in Endocrinology: Approaches to molecular genetic diagnosisin the management of differences/disorders of sex development (DSD): position paper EU COST Action BM 1303 “DSDnet”. Eur J Endocrinol. 2018.
- McPhaul MJ, Marcelli M, Zoppi S, Wilson CM, Griffin JE, Wilson JD. Mutations in the ligand-binding domain of the androgen receptor gene cluster in two regions of the gene. J Clin Invest. 1992; 90(5): 2097.
- Hellmann P, Christiansen P, Johannsen TH, Main KM, Duno M, Juul A. Male patients with partial androgen insensitivity syndrome: a longitudinal follow-up of growth, reproductive hormones and the development of gynaecomastia. Arch Dis Child. 2012 May; 97(5): 403-409.
- Verp MS, Simpson JL: Abnormal sexual differentiation and neoplasia. Cancer Genet Cytogenet 1987; 25: 191.
- Muller J: Morphometry and histology of gonads from twelve children and adolescents with the androgen insensitivity syndrome. J Clin Endocrinol Metab 1984; 59: 785.
- Deans R, Creighton SM, Liao LM, Conway GS. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome CAIS: patient preferences and clinical evidence. Clin Endocrinol Oxf. 2012 Jun; 76(6): 894-898.
- Cools M, Wolffenbuttel KP, Hersmus R, Mendonca BB, Kaprová J, Drop SLS, Stoop H, Gillis AJM, Oosterhuis JW, Costa EMF, Domenice S, Nishi MY, Wunsch L, Quigley CA, T’Sjoen G, Looijenga LHJ. Malignant testicular germ cell tumors in postpubertal individuals with androgen insensitivity: prevalence, pathology and relevance of single nucleotide polymorphism-based susceptibility profiling. Hum Reprod. 2017 Dec 1; 32(12): 2561-2573.
- Weidler EM, Linnaus ME, Baratz AB, Goncalves LF, Bailey S, Hernandez SJ, Gomez-Lobo V, van Leeuwen K. A Management Protocol for Gonad Preservation in Patients with Androgen Insensitivity Syndrome. J Pediatr Adolesc Gynecol 2019 Dec; 32(6): 605-611.
- Khorashad BS, Aghili Z, Kreukels BPC, Reid AG, Roshan GM, Hiradfar M, Talaei A, Cohen Kettenis PT. Mental Health and Disorders of Sex Development/Intersex Conditions in Iranian Culture: Congenital Adrenal Hyperplasia, 5-α Reductase Deficiency-Type 2, and Complete Androgen Insensitivity Syndrome. Arch Sex Behav. 2018 May; 47(4): 931-942.