Glucogenosis hepática como manifestación de síndrome de Mauriac en el adulto
Gonzalo Rojas G1*, Claudio Reinoso A2, María José Valenzuela Pérez3.
Hepatic glycogenosis as a manifestation of Mauriac’s syndrome in adults
- Médico Cirujano. Becado Medicina Interna Universidad Valparaíso. Valparaíso, Chile.
- Médico Internista. Hospital Carlos Van Buren. Valparaíso, Chile.
- Diabetóloga y Nutrióloga. Médico Internista. Hospital Carlos Van Buren. Valparaíso, Chile.
*Correspondencia:
Gonzalo Rojas G. /
gonzalorojasgallegos@gmail.com
Recibido: 16-10-2023.
Aceptado: 23-11-2023.
Resumen: El síndrome de Mauriac es una complicación poco frecuente de la diabetes mellitus tipo 1 mal controlada que se presenta comúnmente en adolescentes con retraso del crecimiento, retraso de la pubertad, rasgos cushingoides, hipercolesterolemia y hepatomegalia. Sin embargo, la única característica de presentación del síndrome de Mauriac puede ser la glucogenosis hepática tanto en adultos como en niños. El pilar del tratamiento de la glucogenosis hepática es el control estricto de los niveles de glucosa, con un excelente pronóstico con un mejor control glucémico. Presentamos un caso de una mujer de 20 años con antecedente de diabetes mellitus tipo 1 (DM1), dislipidemia mixta con mal control metabólico secundario a mala adherencia al tratamiento con múltiples hospitalizaciones por cetoacidosis diabética (CAD). En su última hospitalización se pesquisa hepatomegalia con elevación de enzimas hepáticas asociado a talla baja, rasgos cushingoides y retraso de madurez sexual por lo que se plantea diagnóstico de síndrome de Mauriac.
Palabras clave: Diabetes mellitus tipo 1; Glucogenosis hepática; Hepatomegalia Síndrome de Mauriac.
Abstract: Mauriac syndrome is a rare complication of poorly controlled type 1 diabetes mellitus that commonly presents in adolescents with growth retardation, delayed puberty, Cushingoid features, hypercholesterolemia, and hepatomegaly. However, the only presenting feature of Mauriac syndrome may be hepatic glycogenosis in both adults and children. The mainstay of treatment for hepatic glycogenosis is strict control of glucose levels, with an excellent prognosis with better glycemic control. We present a case of a 20-year-old woman with a history of Type 1 Diabetes Mellitus (DM1), mixed dyslipidemia with poor metabolic control secondary to poor adherence to treatment with multiple hospitalizations for diabetic ketoacidosis (DKA). In her last hospitalization, hepatomegaly with elevated liver enzymes associated with short stature, Cushingoid features and delayed sexual maturity was investigated, for which a diagnosis of Mauriac syndrome was proposed.
Keywords: Hepatic glycogenosis; Hepatomegaly; Mauriac syndrome; Type 1 diabetes mellitus.
Introducción
El síndrome de Mauriac es una complicación rara de la DM1 secundaria a un mal control metabólico en la infancia y adolescencia. Se caracteriza por retraso en el crecimiento, en el desarrollo puberal, trastorno del metabolismo del glucógeno determinando una hepatomegalia por depósito de glucógeno denominada glucogenosis hepática y por otro lado complicaciones por hormonas contrarreguladoras generando un síndrome de Cushing1.
Para el diagnóstico durante la edad adulta se requiere un alto índice de sospecha y requiere como pilar fundamental la presencia de la glucogenosis hepática de la cual su estándar de oro es la biopsia hepática.
Los hallazgos claves para el diagnóstico de glucogenosis hepática son niveles elevados de glucosa, hemoglobina glucosilada (HbA1c, lo que demuestra un mal control glucémico a largo plazo) y aminotransferasas (lo que sugiere daño hepático).
El diagnóstico diferencial incluyen investigaciones de enfermedades infecciosas, causas metabólicas (como la enfermedad de Wilson), obstructivas u oncológicas y pruebas hepáticas autoinmunes. El examen ultrasonográfico del hígado es un procedimiento simple y útil para tener información sobre el tamaño y las características del tejido hepático. Desafortunadamente, la hepatopatía por glucógeno no se puede distinguir clínicamente de la enfermedad del hígado graso no alcohólico o la esteatohepatitis no alcohólica mediante la anamnesis, el examen físico o la ecografía.
El estándar de oro para el diagnóstico es la biopsia hepática, considerando que la preparación del tejido es muy importante para la identificación del glucógeno en los cortes de tejido. Las principales características histológicas de la hepatopatía por glucógeno son una marcada acumulación de glucógeno que conduce a hepatocitos pálidos e hinchados, cambio graso leve o nulo, inflamación mínima o nula, necrosis lobulillar irregular nula o mínima y arquitectura intacta sin fibrosis significativa2.
Caso clínico
Presentamos el caso de una paciente de sexo femenino de 20 años con antecedentes de dislipidemia mixta, DM1 debut a los 3 años múltiples hospitalizaciones por CAD, retinopatía diabética proliferativa con deterioro severo de la visión y calidad de vida. Desde el punto de vista psiquiátrico se describe desde la infancia trastorno de personalidad definido inicialmente como oposicionista desafiante lo cual conllevaba un abandono frecuente a la terapia insulínica, actualmente en controles irregulares por salud mental con sospecha de trastorno afectivo bipolar.
Consulta en urgencia por 2 días de dolor punzante EVA 9/10 en cuadrante superior derecho iniciado de manera progresiva e irradiado en faja asociado a nausea, cefalea.
Al examen físico general destacó febrícula, baja estatura (156 cm de altura), 52 kg de peso corporal y su IMC era de 21.5 kg/m2, caracteres sexuales secundarios retrasados para su edad, facie de luna llena. Al examen segmentario abdomen distendido, hígado 2,5 cm bajo borde costal, con puño percusión positiva a derecha. Dentro de los exámenes destacó elevación de parámetros inflamatorios, HbA1c 12.1%, elevación de transaminasas GOT 506 U/L, GPT 179 U/L, BT 0,2 mg/dL, BD 0,1 md/dL GGT 354 U/L FA 210. U/L, CT 448, HDL 23, TG 1520 y examen de orina inflamatorio.
Tomografía computada (TAC) sin contraste que evidencia moderada hepatomegalia de 18 cm en su eje longitudinal (Figura 1).
Se decide hospitalizar con los diagnósticos de pielonefritis aguda, hepatopatía en estudio, talla baja y sd de Cushing. Para estudiar hepatopatía se solicitó Serología para VIH, virus hepatitis A, B y C negativos, ANA, ASMA, Anti LKM negativos y cinética de fierro compatible con ferropenia. Se descarta Sd de Cushing con cortisol libre urinario 27 ug/24 hrs (VN 4-176)
Al interrogatorio dirigido refiere retraso puberal con adrenarquia a los 14 años, menarquia a los 16 años en comparación a madre, hermana y tías a los 12 años. Estatura materna 170 cm, sin datos de estatura paterna, pero al menos mayor a 170 cm configurando una talla diana mínima de 165 cm.
Durante estadía hospitalaria paciente se logra compensación metabólica logrando disminuir hasta normalizar enzimas hepáticas con hepatomegalia en disminución.
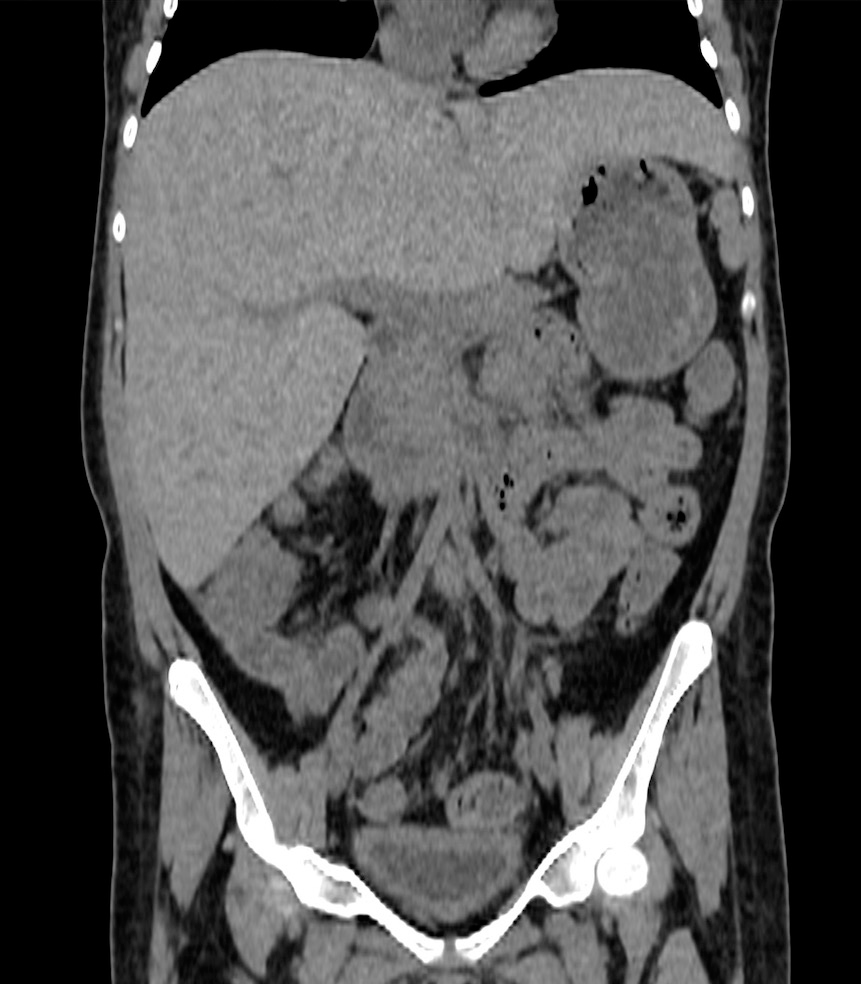 Figura 1: Corte coronal de escáner sin contraste.
Figura 1: Corte coronal de escáner sin contraste. Discusión
Ya han pasado 93 años desde que Leonard Pierre Mauriac en su trabajo titulado “Gros Ventre, Hepatomegalie, Troubles de Las Croissance Chez Les Enfants Diabetiques Traits Depuis Plusieurs Annes Par L’insuline” describiera un síndrome caracterizado por falta de crecimiento, hepatomegalia, enzimas hepáticas elevadas y glucogenosis hepática relacionado con una DM1 mal controlada3.
La glucogenosis hepática puede ser la única característica de presentación del síndrome de Mauriac tanto en adultos como en niños que se desarrolla debido a la acumulación excesiva de glucógeno en los hepatocitos y se caracteriza por hepatomegalia y transaminasas hepáticas elevadas4.
Esta complicación se ve más frecuente en adolescentes sin prevalencia por género y ha disminuido su incidencia gracias a los avances tecnológicos en el control y tratamiento de la diabetes. En general, la glucogenosis hepática (GH) tiene buen pronóstico y rápida resolución tras un adecuado control glucémico, sin progresión a hepatopatía terminal5.
Aunque los mecanismos subyacentes por los que se desarrolla la GH aún no se han dilucidado por completo, las amplias fluctuaciones en las concentraciones de glucosa e insulina parecen ser esenciales para su patogenia. Los niveles de glucosa e insulina en sangre a menudo fluctúan en pacientes diabéticos con mal control metabólico, lo que promueve la acumulación de glucógeno hepático. Los niveles elevados de glucosa en plasma provocan una entrada de glucosa independiente de la insulina en los hepatocitos por difusión facilitada. En el citoplasma de los hepatocitos, la glucosa se convierte irreversiblemente en glucosa-6-fosfato por acción de la glucokinasa, una enzima regulada por la glucosa y la insulina. Luego, la glucosa-6-fosfato se convierte en glucógeno por la glucógeno sintasa, que se convierte de la forma inactiva a la forma activada por una fosfatasa. Esta fosfatasa juega un papel clave en la regulación de este paso de la vía: su concentración la mantiene la insulina, y su actividad depende de la presencia de glucosa. Por lo tanto, la síntesis de glucógeno hepático es consecuencia de la combinación de niveles elevados de glucosa en sangre (que favorecen el flujo de glucosa hacia los hepatocitos) y la hiperinsulinemia (que estimula la conversión de glucosa en glucógeno). Esta situación se observa con frecuencia en pacientes con diabetes inestable que son tratados con insulina por hiperglucemia marcada o prolongada4.
Por primera vez, se informó una asociación genética en un paciente con síndrome de Mauriac en 2016. Describieron una mutación en la subunidad catalítica del complejo enzimático de glucógeno fosforilasa quinasa hepática [mutación PHKG2 G-A en el exón 9] en un paciente con síndrome de Mauriac que desarrolló falla de crecimiento y hepatomegalia masiva. Este complejo enzimático activa la glucógeno fosforilasa que cataliza el primer paso en la degradación del glucógeno. Se demostró que la subunidad mutante actúa de manera dominante para inhibir por completo la actividad de la enzima fosforilasa cinasa de glucógeno y que esto interfiere con la glucogenólisis y provoca un aumento de los niveles de glucógeno en las células hepáticas humanas6.
Volviendo al caso clínico nuestra paciente tiene las características clásicas del síndrome de Mauriac: baja estatura, retraso en la madurez sexual, facies de luna llena y hepatomegalia. Presenta periodos de hiperglucemia no controlada en domicilio con la insulina basal bolo y corrección rápida de la glucosa con infusión de insulina intravenosa en el hospital conduciendo la glucosa al hepatocito (estimulación de la glucógeno sintasa) que se convierte en glucógeno y se deposita en el hígado. Una vez que es dado de alta de regreso a su domicilio, la insulina insuficiente inhibe la glucogenólisis (inhibición de la glucógeno fosforilasa), y el contenido de glucógeno de los hepatocitos aumenta acumulativamente, lo que da como resultado una glucogenosis hepática. El control glucémico deficiente debido a la hipoinsulinemia conduce a la lipólisis y la liberación de cetonas. La cetosis activa la síntesis de cortisol, lo que promueve la liberación de ácidos grasos y la hiperglucemia.
Referencias
- Thakkar, Umang G., Aruna V. Vanikar, and Hargovind L. Trivedi. “Revisit of a Rare Complication of Type 1 Diabetes Mellitus: Mauriac Syndrome.” Practical Diabetes 2017; 34(4): 132-134.
- Giordano, Stefania, Antonio Martocchia, Lavinia Toussan, Manuela Stefanelli, Francesca Pastore, Antonio Devito, Marcello G. Risicato, Luigi Ruco, and Paolo Falaschi. “Diagnosis of Hepatic Glycogenosis in Poorly Controlled Type 1 Diabetes Mellitus.” World Journal of Diabetes. 2014; 5(6): 882-888.
- Mauriac, P. “Gros Ventre, Hepatomegalie, Troubles de Las Croissance Chez Les Enfants Diabetiques Traits Depuis Plusieurs Annes Par L’insuline.” Gax Hebd Med Bordeaux. 1930; 26: 402-410.
- Julián, María Teresa, Núria Alonso, Isabel Ojanguren, Eduarda Pizarro, Enric Ballestar, and Manel Puig-Domingo. “Hepatic Glycogenosis: An Underdiagnosed Complication of Diabetes Mellitus?” World Journal of Diabetes. 2015; 6(2): 321-325.
- Nayak, Samar Pratim, Somashekara Hosaagrahara Ramakrishna, Bargavi Jyothinagaram Sivaprakas, and Subramanian Kannan. n.d. “Mauriac Syndrome: A Rare Cause of Massive Hepatomegaly”. International Journal of Diabetes in Developing Countries. 2021; 41: 697-699.
- MacDonald, Michael J, Noaman M. Hasan, Israr-Ul H. Ansari, Melissa J. Longacre, Mindy A. Kendrick, and Scott W. Stoker. “Discovery of a Genetic Metabolic Cause for Mauriac Syndrome in Type 1 Diabetes.” Diabetes. 2016; 65(7): 2051-259.