Regiones polimórficas del gen 11ß-hidroxiesteroide deshidrogenasa tipo 1 (11ßHSD1) en hipertensión arterial esencial. Posible rol etiopatogénico
Mauricio A. Morales1a, Cristián A. Carvajal1c, Eugenia Ortiz1, Lorena M. Mosso1, Rocío A. Artigas1b, Gareth I. Owen2c,
Carlos E. Fardella1.
Polymorphic regions of 11ß-hydroxysteroid dehydrogenase type 1 (11ßHSD1) in essential hypertension. Possible etiopathogenic role
1Departamento de Endocrinología y
2Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile. Santiago, Chile.
aEstudiante de Bioquímica, Pontificia Universidad Católica de Chile.
bBióloga.
cBioquímico.
Correspondencia a: Dr. Carlos E. Fardella. Departamento de Endocrinología y Metabolismo, P. Universidad Católica de Chile. Lira 85, 5° piso, Santiago, Chile. e mail:cfardella@med. puc.cl
Apoyo financiero: Proyectos Fondecyt 1040834 y 1070876.
Background: Cortisol has been implicated in hypertension and lately reported to be regulated at the pre-receptor level by the 11ßHSD1 enzyme, which converts cortisone (E) to cortisol (F). Over expression of this enzyme in adipose tissue could determine an increase in available cortisol that interacts with the mineralocorticoid receptor (MR) in renal, brain and heart tissue, leading to similar hypertensive effects as in 11ßHSD2 impaired patients. Several polymorphisms have been reported in HSD11B1 gene (CA15, CA19 and InsA83557), which could modify HSD11B1 gene expression or activity. Aim: To determine the distribution and prevalence of CA15, CA19 and InsA83557 in the HSD11B1 gene, and to correlate these results with biochemical parameters in cortisol/ACTH (HPA) and renin-angiotensin-aldosterone (RAA) axis in patients with essential hypertension (EH). Patients and Methods: We studied 113 EH patients (76 non-obese and 37 obese, with a body mass index >30 kg/m2) and 30 normotensive adults (NT). In each patient, we measured serum levels of F, E, serum aldosterone (SA), plasma renin activity (PRA), adrenocorticotrophic hormone (ACTH), the urinary free cortisol/creatinine (UFF/Cr), F/ACTH and SA/PRA ratios. Each polymorphism was studied by PCR and 8% polyacrylamide gel electrophoresis. Statistical associations were evaluated by Pearson correlations and the genetic equilibrium by the Hardy-Weinberg (H-W) equation. Results: We found all three polymorphisms in the EH and the NT group, both in genetic equilibrium. In obese essential hypertensives, the CA15 polymorphism showed association with SA/PRA ratio (r =0.189, p =0.012) and F/ACTH (r =0.301, p 0.048); CA19 also showed correlation with F/ACTH in obese EH (r =-0.220, p =0.009). The InsA83557 polymorphism correlated with UFF/Cr in both EH (r =0.206; p =0.03), and in obese EH (r =0.354; p =0.05). Conclusions: The CA15 and CA19 polymorphism correlated with changes in biochemical parameters in HPA and RAA axis of obese essential hypertensives. These changes may result of modifications in the expression of 11ßHSD1, leading to increased cortisol and aldosterone levels independent of ACTH and renin control, respectively (Rev Méd Chile 2008; 136: 701-10).
(Key words: 11-beta-Hydroxysteroid dehydrogenase type 1; Hypertension; Polymorphism, genetic)
La hipertensión arterial (HTA) constituye una de las patologías más relevantes y con mayor impacto en la salud de la población. Sin embargo, la mayor parte de las veces, la HTA no reconoce una etiología conocida, por lo que se le denomina esencial. Un subtipo de HTA esencial es la HTA hiporreninémica, que se estima correspondería a 20%-30% de la población de hipertensos. En la HTA hiporreninémica se ha planteado que la supresión de renina y la elevación de la presión arterial podrían explicarse a través de una síntesis excesiva de mineralocorticoides.
El hiperaldosteronismo primario es la principal manifestación conocida de la excesiva producción de mineralocorticoides. En la actualidad, se sabe que esta enfermedad da cuenta de aproximadamente el 50% de los casos de HTA hiporreninémica, a través de una mayor reabsorción de sodio y agua a nivel renal, generando hipervolemia y, secundariamente, el aumento de las cifras tensionales y la supresión de renina1. En el pasado, estas cifras eran significadamente más bajas porque el screening de esta enfermedad se basaba en determinaciones de potasio plasmático, el cual sólo se encuentra disminuido en alrededor del 20% de los casos, lo que explicaba el subdiagnóstico2.
El cortisol es otro esteroide que puede ejercer efecto prohipertensivo. En la literatura existe evidencia de cómo el cortisol juega un rol importante en el control de la presión arterial induciendo hipertensión en el 70% de los pacientes con Síndrome de Cushing y en un alto número de pacientes bajo tratamiento con corticoides3. Además, en un reciente estudio, nuestro grupo demostró la existencia de una correlación negativa entre los niveles de cortisol libre urinario (CLU) y actividad renina plasmática (ARP), como también de CLU y aldosterona plasmática (AP), sugiriendo que cortisol (F) favorecería un mecanismo prohipertensivo activando la reabsorción de Na+/H2O a nivel renal vía receptor de mineralocorticoides (MR), con un efecto semejante al descrito para la aldosterona4.
Las 11ß-hidroxiesteroide deshidrogenasas tipo 1 y 2 constituyen el tercer factor involucrado en el control de la presión arterial y génesis de la HTA hiporreninémica. La más conocida es la tipo 2, denominada 11ßHSD2, localizada preferentemente en riñón y colon, encargada de convertir el cortisol a cortisona y con ello impedir la unión del cortisol al MR. Esta enzima ha sido ampliamente estudiada por nuestro grupo y hemos demostrado qué defectos parciales o totales de esta enzima provocan la deficiente inactivación de este metabolito, lo que promueve la activación del MR, generando una hipertensión volumen-dependiente5.
La enzima 11ß-hidroxiesteroide deshidrogenasa tipo 1 (11ßHSD1), a diferencia de la tipo 2, es expresada preferentemente en el hígado, tejido adiposo, vasos sanguíneos y cerebro6. Esta enzima, producto de su actividad hidroxiesteroide reductasa, metaboliza in vivo la cortisona inactiva a cortisol6,7. El cortisol se uniría rápidamente al receptor de glucocorticoides. Modelos animales que sobreexpresan el gen de esta enzima, HSD11B1, presentan fenotipos de hipertensión y aumento de la aldosterona, sugiriendo que la enzima 11ßHSD1 podría estar modulando el eje renina-angiotensina-aldosterona8,9. Ratones obesos, por otra parte, muestran una gran actividad hidroxiesteroide reductasa, lo que implica una alta expresión de 11ßHSD110. Por otro lado, animales knockout para HSD11B1 presentan hiperplasia adrenal y aumento del cortisol plasmático como efecto compensatorio a la falta de regeneración del cortisol11.
Se han descrito en la literatura distintos polimorfismos del gen HSD11B1. Los más relevantes resultan ser: una inserción de A en el intrón 3 (InsA83557) y dos microsatélites dinucleotídicos (CA15 y CA19) ubicados en el intrón 4. Estas repeticiones se denominan así por presentar 15 y 19 repeticiones de CA en su secuencia, respectivamente (Figura 1). Su relevancia radica en que podrían alterar la expresión génica del gen HSD11B1. Estudios en este campo se han destinado a elucidar un posible rol de estas secuencias repetitivas en la contribución genética en enfermedades complejas, como síndrome metabólico y obesidad central8,12.
Por su parte, el polimorfismo (inserción de adenina) que ocurre en la posición nucleotídica 83557 (InsA83557) del intrón 3, es un polimorfismo presente en distintas poblaciones y se ha intentado establecer su asociación con ciertas patologías13, una de ellas el Síndrome de Ovario Poliquístico (SOP), uno de los tantos fenómenos observados en pacientes con Síndrome Metabólico14. De esta forma, se ha intentado asociar los microsatélites y el polimorfismo InsA en el desarrollo de patologías de alta prevalencia, como la obesidad. En ese sentido, no se ha encontrado asociación entre los polimorfismos con el IMC, pero sí con la razón cintura-cadera, que es una medida antropométrica para evaluar la distribución de grasa a nivel del perímetro de cintura. Esta variable se ha transformado en un buen indicador clínico del grado de adiposidad visceral y de la presencia de complicaciones metabólicas asociadas15,16. En obesidad visceral o androgénica, se ha demostrado que existe un aumento en la actividad de 11ßHSD116, lo que repercute en el grado de diferenciación de los adipocitos, que sería inducida por cortisol17. Asimismo, se ha podido demostrar una asociación entre el microsatélite CA15 y una mayor actividad in vivo de la enzima 11ßHSD1, determinada por la razón (allo-THF+THF)/THE15.
Es posible plantear que una sobreexpresión de esta enzima determine un aumento en la generación del cortisol y secundariamente desarrolle efectos pro-hipertensivos (por ejemplo, activación del receptor de mineralocorticoides) y, con ello, desarrollo de hipertensión arterial. Las variantes alélicas de estos polimorfismos podrían determinar cambios en la transcripción del gen HSD11B1 y, con ello, modificaciones en la actividad de la enzima que podrían explicar la aparición de hipertensión y obesidad. El objetivo de este trabajo es determinar la distribución y prevalencia de los polimorfismos descritos en el gen HSD11B1, y evaluar su asociación con cambios en el eje ACTH-cortisol y renina-angiotensina-aldosterona en hipertensos esenciales obesos y no obesos que podrían estar indicando cambios en la actividad de 11ßHSD1.

Pacientes y Métodos
Pacientes. Se estudiaron 113 pacientes hipertensos esenciales (HE) provenientes de programas de atención a hipertensos crónicos de un centro de atención primaria de la comuna de Puente Alto (Consultorio Padre Villaseca). También se estudiaron 30 sujetos sanos provenientes de la misma población, como controles normotensos. Se consideraron hipertensos aquellos pacientes cuya presión era determinaciones separadas por una semana de intervalo. Se excluyeron pacientes con presencia de patologías crónicas que pudiesen afectar la medición de renina o que provoquen HTA secundaria, tales como insuficiencia renal, insuficiencia cardíaca, daño hepático crónico o endocrinopatías, y aquellos que hubieran recibido tratamientos con corticoides. Se consideraron normotensos quienes presentaban cifras de PA <140/85 mmHg, determinadas en 3 ocasiones. Se consideraron obesos a aquellos pacientes cuyo IMC fuera mayor a 30 kg/m2. Las características clínicas de HE (obesos y no obesos) y normotensos (NT) se presentan en la Tabla 1. A todos los pacientes y voluntarios se les solicitó el consentimiento informado para participar en el estudio de acuerdo a las normas de la Declaración de Helsinki y el protocolo fue aprobado por el Comité de Ética de la Dirección de Investigación de la Escuela de Medicina de la Pontificia Universidad Católica de Chile.
A cada paciente que ingresó a nuestro estudio se le extrajo una muestra de sangre entre las 08:00 y 10:00 AM, después de un ayuno de 12 h. Todos ellos se encontraban con una dieta normosódica y dos semanas previas al estudio se les cambió la terapia antihipertensiva por fármacos que no afectaban el eje renina-angiotensina-aldosterona19,20. Al ingreso se les controló peso y talla. Posteriormente se les colocó un catéter en la vena antecubital y se les dejó en reposo (sentado) por 10 min, después de lo cual se tomaron muestras de sangre para medir niveles de cortisol (F), cortisona (E), ACTH, los niveles de actividad de renina plasmática (ARP), de aldosterona (AP), glicemia, colesterol total, LDL, HDL, triglicéridos, cortisol libre urinario normalizado por creatinina (CLU/Cr).
Métodos. La ARP fue determinada por el método previamente descrito por Menard21 y su valor normal oscila entre 1-2,5 ng/ml*h, operacionalmente, se consideraron niveles bajos de ARP cuando éstos eran <0,5. El límite inferior de detección de la ARP es 0,1 ng/ml*h22. El coeficiente de variación intra e interensayo fue de 6,1% y 8,2%, respectivamente. La AP fue medida por un kit comercial de Diagnostic Products Corp (Los Angeles, California, USA), cuyo valor normal es entre 1-16 ng/dL. El coeficiente de variación intra e interensayo para la AP fue de 5,1% y 7,1%, respectivamente. La determinación de F y E se hizo con un ensayo de RIA desarrollado en nuestro laboratorio4. La determinación de excreción urinaria de creatinina (Cr) se determinó por la técnica Jaffe23. La glucosa en ayuno fue determinada por método de glucosa hexoquinasa (Hitachi). Triglicéridos y colesterol fueron determinados por un ensayo enzimático-colorimétrico (Hitachi). ACTH fue determinado con un ensayo inmunométrico quimioluminiscente (Diagnostic Products Corp.).
Genotipificación de variantes alélicas. Se aisló el ADN
genómico a partir de leucocitos de sangre periférica de
pacientes y controles usando DNAzol (Invitrogen, USA).
En todos ellos se tipificó la región microsatélite según el
número de repeticiones CA y se determinó el genotipo de
la inserción InsA83557 mediante RFLP, usando la enzima
XcmI (New England Biolabs, USA). El diseño de los partidores
se realizó en nuestro laboratorio de forma automatizada
mediante el software Primer3 (http://frodo.wi.mit.
edu/cgibin/primer3 /primer3_www.cgi).
Las reacciones de PCR se realizaron en un termociclador Mastercicler® Eppendorf usando aproximadamente 0,1 a 0,5 μg de ADN, 5 pmoles del partidor sentido, 5 pmoles de partidor antisentido, cuyas características se encuentran en la Tabla 2, 1,25 pmoles de cada dNTP y 1 U de ADN Taq polimerasa en Tris HCl 75 mM (pH 9), KCl 50mM, (NH4)2 SO4 20 mM y MgCl2 2 mM en un volumen final de 25 μL. Las secuencias y propiedades de los partidores se encuentran en la Tabla 3. El programa térmico que se usó para la PCR constó de las siguientes etapas: 1) una desnaturación inicial a 94°C durante 2 min; 2) 30 ciclos de amplificación con una desnaturación a 94°C durante 30 s, un apareamiento a 55°C, tanto para InsA83557 como para los microsatélites CA15 y CA19, durante 30 s y una polimerización a 72°C durante 30 s; y 3) una polimerización final durante 10 min a 72°C. El RFLP se realizó con el mismo termociclador, utilizando 10 μL del producto del PCR, equivalentes aproximadamente a 10 μg, 0,13 U de XcmI en 50 mM NaCl, 10 mM Tris-HCl (pH9), 10 mM MgCl2 y 1 mM DTT en un volumen final de 25 μL. El programa térmico para la digestión enzimática fue el siguiente: digestión enzimática por 2 h a 37°C y una inactivación por calor durante 20 min a 65°C.
Identificación de los alelos. Los productos de la PCR se sometieron a una electroforesis en un gel de poliacrilamida (19:1) al 8% durante 4 h a 150 volts constantes. Luego el gel se tiñó en una solución de bromuro de etidio 0,45 μg/mL y el resultado se visualizó en un transiluminador de luz UV. Para la región microsatélite CA15 y CA19 se comparó el tamaño de las bandas correspondientes al amplicón que contiene la región microsatélite de la respectiva repetición según su migración electroforética, usando estándares de peso molecular conocido e individuos cuyo largo de las regiones se conoció mediante secuenciación automática.
Secuenciación de las regiones polimórficas. Se secuenció la zona de la repetición CA15 y CA19 en 3 controles respectivamente, de manera de conocer el número exacto de repeticiones que ellos tenían y poder usarlos como control en la identificación de los alelos mediante electroforesis. Los fragmentos a secuenciar se amplificaron mediante una PCR que se realizó como se describió anteriormente. Cada uno de los amplicones se purificó con el sistema de purificación “Qiaquick gel extraction kit” (Qiagen, USA). La secuenciación se realizó mediante un secuenciador automático ABI prism-377 DNA Sequencer (Applied Biosystem, USA). Las secuencias nucleotídicas de cada paciente se compararon con la del gen HSD11B1, ubicado en el cromosoma 1 (GI:32455237), mediante el programa BLAST24.
Estadistica. En base al analisis de frecuencias alelicas para las regiones microsatelites y genotipicas para InsA83557, se tabulo la distribucion de tales polimorfismos en las poblaciones estudiadas. En hipertensos y normotensos se realizo el analisis de Hardy-Weinberg (H-W)23. Se consideraron estadisticamente significativos valores de significancia p <0,05. Se realizaron pruebas de χ2 para el analisis de frecuencias alelicas y ANOVA para analizar las diferencias entre parametros bioquimicos entre los grupos de HE y NT. El analisis estadistico se llevo a cabo mediante el programa Minitab14 (Minitab Inc. USA). Se realizaron correlaciones de Pearson o Spearman uni y multivariadas entre el largo de los alelos de las variantes alelicas CA15 y CA19, ademas del genotipo presentado por InsA83557, respecto de las variables clinicas y bioquimicas: IMC, F, E, ACTH, AP, ARP, glicemia, CLU/Cr, colesterol total, HDL, LDL, y las razones F/ACTH y AP/AR.
Tabla 1. Parámetros clínicos y bioquímicos de pacientes hipertensos esenciales (HE) y sujetos normotensos (NT*).
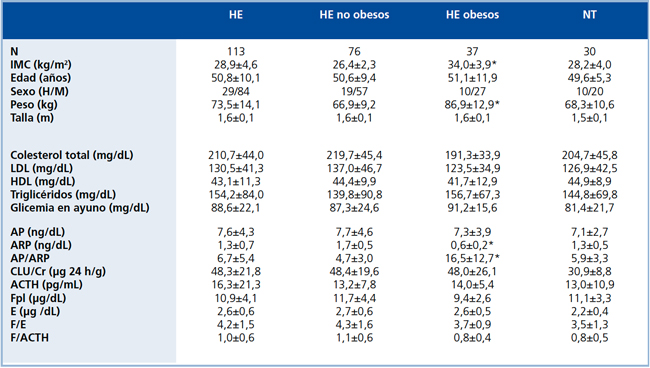
El grupo HE se subdividió en HE esenciales obesos y no-obesos, definiendo a pacientes obesos como aquellos con IMC>30 kg/m2. Valores se encuentran expresados en promedio ± DS.
*Parámetro significativamente diferente entre los grupos.
Tabla 2. Partidores sentido y antisentido utilizados en la amplificación por PCR de polimorfismos CA15, CA19 e InsA83557 del gen HSD11B1.

Tabla 3. Frecuencias genotípicas (F) de las regiones microsatélites, CA15 y CA19 y de InsA83557 del gen HSD11B1 en pacientes HE, obesos y no obesos y sujetos normotensos (NT).
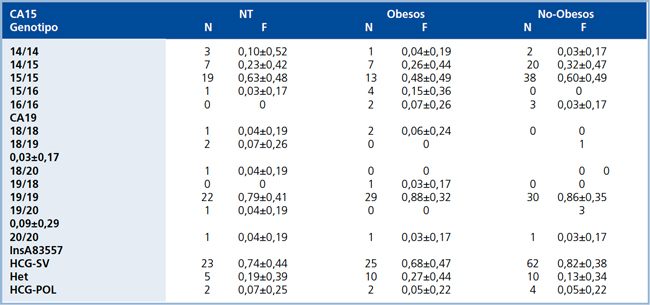
Se definen a pacientes obesos como aquellos con IMC >30 kg/m2. Valores se encuentran expresados en frecuencias ± DS. HCGSV: Homocigoto silvestre. Het: Heterocigoto. HCG-POL: Homocigoto polimórfico.
Resultados
Identificacion y genotipificacion de las zonas microsatelites, CA15 y CA19, e InsA83557. En HE y normotensos se detectaron tres variantes alelicas diferentes; para CA15 se detectaron 14, 15 y 16 repeticiones; para C19 se detectaron 18, 19 y 20 repeticiones. InsA83557 presento a su vez tres genotipos distintos: homocigoto silvestre (HCGSV), heterocigoto (HET) y homocigoto polimorfico (HCG-POL). En HE las regiones microsatelites cumplian con el equilibrio de H-W, tanto en el subgrupo obesos como en HE noobesos para CA15 (χ2 =1,129; 3 GL; p =0,77 y χ2 =3,236; 3 GL;p =0,357, respectivamente) para C19 (χ2 =4,27; 4 GL; p =0,371 y χ2 =1,218; 3 GL; p =0,128, respectivamente) y para la InsA83557 (χ2 =1,85; 2GL; p =0,404 y χ2 =0,164; 2 GL; p=0,921, respectivamente). En normotensos, el CA15 presento equilibrio H-W (χ2 =6,563; 3 GL; p =0,083), a diferencia del CA19 y la InsA83557 que no lo presentaron (χ2 =26,298; 5 GL; p <0,001 para CA19; χ2 =17,885; 2 GL; p <0,001 para InsA83557). Las frecuencias genotipicas de las tres regiones variables se presentan en la Tabla 3, mientras que las frecuencias alelicas de los microsatelites se presentan en la Tabla 4.
Relacion entre las variantes alelicas y parametros bioquimicos estudiados. El microsatelite CA15 en el grupo total de HE y normotensos no se asocio con ninguna de las variables bioquimicas estudiadas. Sin embargo, en HE obesos se observo una asociacion entre el largo del alelo con AP/ARP (r =0,189; p =0,012) y con F/ACTH (r =0,301; p =0,048).
El microsatelite CA19 en el grupo total de HE mostro asociacion con el IMC (r =-0,220; p=0,009), en normotensos no se detecto ninguna asociacion con las variables bioquimicas estudiadas. En HE obesos se observo una asociacion negativa entre el largo del alelo con F/ACTH (r =-0,294; p =0,025).
La presencia de InsA83557 presento una asociacion con el CLU/Cr en el grupo total de HE (r =0,206; p =0,03) y en el subgrupo de obesos en grupo total (r =0,354; p =0,05). En normotensos no se demostro ninguna correlacion con este polimorfismo.
Tabla 4. Frecuencias alélicas de cada variante de las regiones microsatélites, CA15 y CA19, del gen HSD11B1 presentes en HE, obesos y no obesos y en sujetos normotensos (NT).
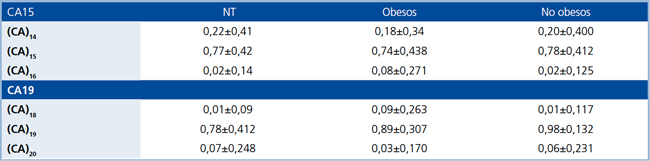
Se definen a pacientes obesos como aquellos con IMC >30 kg/m2. Valores se encuentran expresados en promedio ± DS.
Discusion
Los resultados de este trabajo muestran la existencia de variantes alelicas de los microsatelites CA15 y CA19 y la insercion InsA83557 en el gen HSD11B1 en poblacion HE y normotensa chilena. El analisis de correlacion de las distintas variantes de estos polimorfismos con variables bioquimicas demostro una asociacion de estas con cambios en los ejes renina-angiotensina y la razon F/ACTH, siendo mas evidentes en pacientes obesos.
Estos cambios serian inducidos por modificaciones locales en la expresion de la enzima 11sHSD114,26, que determinarian el aumento en los niveles de cortisol (F) y un desbalance en la regulacion de aldosterona.
La asociacion observada entre la variable F/ACTH y las variantes alelicas de los polimorfismos CA15 y CA19 en pacientes HE obesos podria dar cuenta de una mayor sintesis local de cortisol para un nivel dado de ACTH, sugiriendo un probable efecto de estos polimorfismos en la expresion de la enzima 11sHSD1, favoreciendo una mayor conversion de F a partir de E, independiente de la accion de ACTH. Este mecanismo podria ser evaluado ex vivo midiendo las concentraciones de cortisol en tejido graso de los pacientes. Por otra parte, la presencia de InsA83557, ya sea en forma homocigota o heterocigota, se asocio positivamente a un aumento del CLU/Cr, lo que indicaria mayor acumulacion de F en orina, reflejo quizas de una mayor generacion de F a partir de E, tambien secundaria a un aumento de la actividad de la enzima. Asimismo, la mayor disponibilidad de F puede activar distintos mecanismos prohipertensivos, uno de ellos podria ocurrir a nivel de la activacion del MR, contribuyendo a la reabsorcion de sodio a nivel renal. Este ultimo mecanismo pro-hipertensivo es clave en patologias que presentan deficit en la actividad de la enzima 11sHSD227. En nuestros pacientes con HE se logra apreciar un aumento de la relacion F/E en sangre que, aunque no alcanzo diferencias significativas, podria estar indicando un deficit parcial de esta enzima como ha sido comunicado previamente por nuestro grupo3,4.
Los pacientes HE obesos mostraron un aumento de la relacion AP/ARP asociado a la presencia de alelos mas largos de CA15, sin llegar a valores sugerentes de hiperaldosteronismo (HAP). Este hecho ha sido observado en modelos animales que sobreexpresan HSD11B1, donde el cuadro hipertensivo seria secundario a un aumento en la generacion de angiotensinogeno8,9, el cual promueve la estimulacion de aldosterona independiente del control de renina y asi contribuiria a la genesis del cuadro hipertensivo. Sin embargo, de acuerdo a los datos que presentan nuestros pacientes, podríamos considerar este fenómeno como de menor impacto, ya que los valores de aldosterona no son diferentes en los tres grupos y es sólo la renina la que varía, la cual podría estar disminuida por el estado hipervolémico de nuestros pacientes HE.
La razón del porqué estas asociaciones se observan sólo en pacientes HE obesos no es clara, sin embargo, existe evidencia que HSD11B1 aumentaría su expresión en tejido adiposo, como se ha visto en distintos estudios in vitro9,16, siendo mayor la expresión en el tejido adiposo visceral por sobre los otros compartimentos de tejido adiposo. Esta expresión, sin embargo, es tejido-específica: en el tejido adiposo subcutáneo menor nivel de transcripción de HSD11B1 y mayor transcripción del GR, al contrario del tejido adiposo visceral, donde hay gran transcripción de HSD11B1 y baja transcripción del GR16. Modelos de animales obesos presentan una actividad elevada de HSD11B1 en el tejido adiposo visceral en comparación a tejido adiposo subcutáneo y músculo esquelético9. En nuestro estudio, observamos que el acortamiento de CA19 se asocia con un aumento del IMC, mostrando el compromiso de la obesidad en estos pacientes. Esta evidencia, junto con la de otros estudios18,26, sugiere que la obesidad sería un factor determinante en el aumento de la transcripción de HSD11B1 a través de una mayor regeneración local de cortisol, lo que contribuiría a explicar por qué sólo los hipertensos obesos con determinados polimorfismos logran expresar los cambios en la expresión de esta enzima. En este contexto, datos preliminares de nuestro grupo muestran una importante asociación entre cortisol plasmático y razón cintura-cadera (r =-0,328, p =0,007), los que concuerdan con lo reportado en la población del estudio MONICA, donde existe relación además de estos alelos con la razón de los metabolitos de cortisol urinario (THF + allo-THF/THE)13,28.
En resumen, demostramos la presencia de variantes polimórficas en dos regiones microsatélites y una inserción nucleotídica en el gen HSD11B1 en HE chilenos. Estos polimorfismos podrían contribuir a la modificación de la actividad de la enzima 11ßHSD1 y así podría explicar la génesis del cuadro hipertensivo mediante la generación local de cortisol en el tejido adiposo.
Referencias
- Mosso L., Fardella C., Montero J., Rojas P., Sánchez O., Rojas V., et al. Alta Prevalencia de Hiperaldosteronismo Primario no Diagnosticado en hipertensos catalogados como esenciales. Rev Méd Chile 1999; 127: 800-6.
- Mulatero P., Stowasser M., Loh KC., Fardella CE., Gordon RD., Mosso L., et al. Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. J Clin Endocrinol Metab 2004; 89: 1045-50.
- Krall P., Mosso L., Carvajal C., Rojas A., Fardella C. Elevación del cortisol urinario en hipertensos esenciales hiporreninémicos. Rev Méd Chile 2004; 132: 1053-9.
- Krall P., Carvajal C., Ortiz E., Muñoz C., Garrido JL., Mosso L., Fardella C. Urinary free cortisol is not a biochemical marker of hypertension. Am J Hypertens 2007; 20: 459-65.
- . Carvajal C., González A., Romero D., González A., Mosso L., Lagos E., et al. Two homozygous mutations in the 11ß-Hydroxysteroid dehydrogenase type 2 gene in a case of apparent mineralocorticoid excess. J Clin Endocrinol Metab 2003; 88: 2501-7.
- Tomlinson JW., Walker EA., Bujalska IJ., Draper N., Lavery GG., Cooper MS., et al. 11ß-Hydroxysteroid dehydrogenase type 1: A tissuespecific regulator of glucocorticoid response. Endocrine Reviews 2004; 25: 831-66.
- Bujalska IJ., Draper N., Michailidou Z., Tomlinson JW., White PC., Chapman KE., et al. Hexose-6-phosphate dehydrogenase confers oxoreductase activity upon 11ß-hydroxysteroid dehydrogenase type 1. J Mol Endocrinol 2005; 34: 675-84.
- Masuzaki H., Yamamoto H., Kenyon CJ., Elmquist JK., Morton NM., Paterson JM., et al. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest 2003; 112: 83-90.
- Masuzaki H., Paterson J., Shinyama H., Morton NM., Mullins JJ., Seckl JR., Flier JS. A Transgenic Model of Visceral Obesity and the Metabolic Syndrome. Science 2001; 294: 2166-70.
- Livingstone DEW., Jones GC., Smith K., Jamieson PM., Andrew R., Kenyon CJ., Walker BR. Understanding the role of glucocorticoids in obesity: tissuespecific alterations of corticosterone metabolism in obese zucker rats. Endocrinology 2000; 141: 560-3.
- Kotelevtsev Y., Holmes MC., Burchell A., Houston PM., Schmoll D., Jamieson P., et al. 11ß-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci USA 1997; 94: 14924-9.
- Masuzaki H., Flier JS.. Tissue-specific glucocorticoid reactivating enzyme, 11 beta-hydroxysteroid dehydrogenase type 1 (11 beta-HSD1)— a promising drug target for the treatment of metabolic syndrome. Curr Drug Targets Immune Endocr Metabol Disord 2003; (4): 255-62.
- White PC. Genotypes at 11ß-Hydroxysteroid Dehydrogenase Type 11B1 and Hexose-6-Phosphate Dehydrogenase Loci Are Not Risk Factors for Apparent Cortisone Reductase Deficiency in a Large Population- Based Sample. J Clin Endocrinol Metab 2005; 90: 5880-3.
- San Millán JL., Botella-Carretero JI., Alvarez-Blasco F., Luque- Ramírez M., Sancho J., Moghetti P., Escobar-Moncada H. A Study of the Hexose-6- phosphate dehydrogenase gene R453Q and 11ß-Hydroxysteroid dehydrogenase type 1 gene 83557insA polymorphisms in the polycystic ovary syndrome. J Clin Endocrinol Metab 2005; 90:4157-62.
- Draper N., Echwald SM., Lavery GG., Walker EA., Fraser R., Davies E., et al. Association Studies between Microsatellite Markers within the Gene Encoding Human 11ß-Hydroxysteroid Dehydrogenase Type 1 and Body Mass Index, Waist to Hip Ratio, and Glucocorticoid Metabolism. J Clin Endocrinol Metab 2002; 87: 4984-90.
- Michailidou Z., Jensen MD., Dumesic DA., Chapman KE., Seckl JR., Walker BR., Morton NM. Omental 11beta-hydroxysteroid dehydrogenase 1 correlates with fat cell size independently of obesity. Obesity 2007; 15: 1155-63.
- Bujalska IJ., Kumar S., Hewison M., Stewart PM. Differentiation of Adipose Stromal Cells: The Roles of Glucocorticoids and 11ß-hydroxysteroid Dehydrogenase. Endocrinology 1999; 140: 3188-96.
- Bujalska IJ., Quinkler M., Tomlinson JW., Montague CT., Smith DM., Stewart PM. Expression profiling of 11ß-hydroxysteroid dehydrogenase type-1 and glucocorticoid-target genes in subcutaneous and omental human preadipocytes. J Mol Endocrinol 2006; 37: 327-40.
- Seifarth C., Trenkel S., Schobel H., Hahn EG., Hensen J. Influence of antihypertensive medication on aldosterone and renin concentration in the differential diagnosis of essential hypertension and primary aldosteronism. Clin Endocrinol 2002;576: 457-65.
- Mulatero P., Rabbia F., Milan A., Paglieri C., Morello F., Chiandussi L. Drug effects on aldosterone/plasma renin activity ratio in primary aldosteronism. Hypertension 2002; 40: 897-902.
- Menard J., Corvol P. Mesure l’activite rénine plasmatique de l’homme par le dosage radioin-munologique de l’angiotensine I. Tech. Radioinmunol 1972, Paris, Inserm 459.
- Montero J., Soto J., Fardella C., Foradori A., Valdés G. Medición de la actividad renina plasmática: optimización del método. Rev Méd Chile 1998; 126: 151-4.
- Hervey GR. Determination of creatinine by the Jaffe reaction. Nature 1953; 171: 1125.
- Altshul SF., Gish W., Miller W., Myers EW., Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 3215: 403-10. http://www.ncbi. nlm.nih.gov/blast/bl2seq/bl2.html
- . Llorca J, Prieto-Salceda D, Combarros O, Dierssensotos T, Berciano J. Riesgos competitivos de muerte y equilibrio de Hardy-Weinberg en estudios de casos y controles sobre asociación entre genes y enfermedades. Gac Sanit 2005; 19: 321-4.
- Rask E., Walker BR., Söderberg S., Livingstone DEW., Eliasson M., Johnson O. Tissue-Specific Changes In Peripheral Cortisol Metabolism In Obese Women: Increased Adipose 11ß-Hydroxysteroid Dehydrogenase Type 1 Activity. J Clin Endocrinol Metab 2002; 87: 3330-6.
- Carvajal C., Romero DG., Mosso LM., González AA., Montero J., Fardella CE. Biochemical and Genetic Characterization of 11 ß-Hydroxysteroid Dehydrogenase Type 2 In Low-Renin Essential Hypertensives. J Hypertens 2005; 23: 71-7.
- Löwel H., Döring A., Schneider A., Heier M., Thorand B., Meisinger C.; MONICA/KORA Study Group. The MONICA Augsburg surveys—basis for prospective cohort studies. Gesundheitswesen 2005; Suppl 1: S13-8.