Actualización en el manejo del Síndrome de Turner en niñas y adolescentes. Revisión de la Literatura e Incorporación de Recomendaciones de las nuevas Guías Clínicas
Carola Goecke H.1, Hernán García B.1
Update on the management of Turner Syndrome in girls and adolescents. Review of the Literature and Incorporation of Recommendations of the new Clinical Guidelines
1 Unidad de Endocrinología Pediátrica, División de Pediatría, Pontificia Universidad Católica de Chile, Santiago-Chile.
Correspondencia
Carola Goecke Hochberger
Pontificia Universidad Católica de Chile, Escuela de Medicina, División de
Pediatría.
Diagonal Paraguay #362, 8vo piso, Santiago.
Código Postal: 8330074.
Teléfono (56-2) 2354 3402. Fax: 56-226384307
Correo: cmgoecke@uc.cl
Recibido: 07-05-2018
Aceptado: 08-08-2018
Los autores afirman que no recibieron
apoyo financiero alguno para la elaboración
de este manuscrito.
Abstract: Turner syndrome (TS) is a common disorder (1/2.000 women) that affects multiple organs at different stages of life and needs a multidisciplinary approach. It can be present in women of all ethnicities and is caused by a monosomy of the X chromosome that causes a haploinsufficiency of certain genes. Its main features consist of specific but variables physical characteristics, congenital heart defects, renal anomalies, middle and inner ear diseases, skeletal alterations, and from the endocrinological point of view, short stature and ovarian insufficiency. Given the comorbidities associated with TS, it has been estimated that they have an increased risk of mortality (up to 3 times more) and a reduction in life expectancy of approximately 13 years. Depending on the genotype, the abnormalities can become very subtle, in these cases the diagnosis is late, when the adolescent consults, for example, for primary amenorrhea or an adult woman for infertility. Once the diagnosis is confirmed by a karyotype, these patients must remain in pediatric control in a continuous way to investigate associated pathologies in a timely manner, with periodic evaluations by specialists, such as otolaryngologists, cardiologists, neurologists and endocrinologists, among others. Numerous advances in the care of these patients gave rise to new guidelines published in 2017. In this article we will comment on the main conditions associated with TS and its specific etiology, we will mention what is relevant regarding the genotype-phenotype relationship in this syndrome and we will discuss the fundamental aspects of the control of the TS patient, with emphasis on the treatment of short stature and ovarian insufficiency, as well as the cardiovascular aspects and those related to fertility.
Key words: Turner Syndrome; Gonadal Dysgenesis; Sex Chromosome Disorders of Sex Development.
Resumen: El Síndrome de Turner (ST) es una patología frecuente (1/2.000 mujeres) que afecta múltiples órganos en distintas etapas de la vida y necesita un enfoque multidisciplinario. Se produce por una monosomía del cromosoma X que provoca una haploinsuficiencia de determinados genes. Sus características principales consisten en un fenotipo característico pero variable, con presencia de cardiopatías congénitas, anomalías renales, enfermedades del oído medio e interno, alteraciones esqueléticas, y del punto de vista endocrinológico, talla baja e insuficiencia ovárica. Dadas las comorbilidades asociadas al ST, principalmente cardiovasculares (CV), presentan mayor mortalidad con respecto a la población general (hasta 3 veces más). Dependiendo del genotipo, las anomalías pueden llegar a ser muy sutiles, realizándose en estos casos el diagnóstico en forma tardía, cuando la adolescente consulte, por ejemplo, por amenorrea primaria o una mujer adulta por infertilidad. Una vez confirmado el diagnóstico mediante un cariotipo, estas pacientes deben permanecer en control endocrinológico pediátrico en forma continua hasta la transición hacia adultos, con el fin de pesquisar patologías asociadas en forma oportuna. Por ello requieren evaluaciones periódicas por especialistas, tales como otorrinolaringólogos, cardiólogos, neuropsiquiatras, entre otros. Numerosos avances en el cuidado de estas pacientes, dieron origen a nuevas guías publicadas el 2017. En este artículo comentaremos sobre las principales condiciones asociadas al ST y su etiología específica, mencionaremos lo relevante respecto a la relación genotipo-fenotipo en este síndrome y discutiremos los aspectos fundamentales del control de la paciente con ST, haciendo énfasis en el tratamiento de la talla baja y la insuficiencia ovárica, así como los aspectos CV y los relacionados a fertilidad.
Palabras clave: Síndrome de Turner; Disgenesia gonadal; Desorden del Desarrollo Sexual Cromosómico.
El síndrome de Turner (ST) es uno de los desórdenes genéticos más frecuentes, con una incidencia de 1/2.000 recién nacidos (RN) vivos de sexo femenino1. La primera descripción de una niña con estigmas físicos característicos de ST (Figura 1) fue realizada en 1930 por el pediatra alemán Otto Ullrich2; ocho años después el endocrinólogo estadounidense Henry Turner publicó un artículo sobre siete mujeres portadoras de talla baja, cuello alado e infantilismo sexual3. Es una patología multisistémica causada por una anomalía cromosómica determinada por la alteración numérica o estructural del cromosoma X4. Desde el punto de vista endocrinológico, representa una causa importante de talla baja e insuficiencia ovárica en mujeres5.
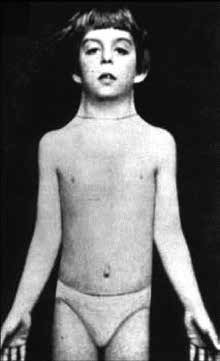
Figura 1. Paciente con ST, descrita originalmente por Otto Ullrich (extraído con autorización de Eur J Pediatr. 1930; 49: 271-276).
| Características Clínicas | Frecuencia (%) |
| Talla Baja | 98 |
| Falla Gonadal | 95 |
| Micrognatia | 60 |
| Cúbito Valgo | 47 |
| Implantación baja del cabello | 42 |
| Cuello Corto | 40 |
| Paladar Ojival | 38 |
| 4° Metacarpiano Corto | 37 |
| Nevos Multiples | 25 |
| Cuello Alado | 25 |
| Linfedema de Manos y Pies | 22 |
| Displasia Ungueal | 13 |
| Escoliosis | 11 |
| Deformidad de Madelung | 7 |
Dentro de los signos clínicos se encuentran el tórax ancho (“en escudo”), cúbito valgo y teletelia, entre otros (Tabla 1). Además de los elementos fenotípicos característicos del examen físico, las manifestaciones incluyen insuficiencia ovárica, pérdida auditiva precoz, defectos cardiacos (coartación aórtica y defectos ventriculares septales), alteraciones renales (agenesia unilateral, duplicación uretral, riñón en herradura), desarrollo neurocognitivo particular y mayor incidencia de enfermedades autoinmunes como hipotiroidismo y enfermedad celiaca6.
El ST se caracteriza por una alta variabilidad fenotípica, fluctuando desde la forma clásica hasta pacientes que son prácticamente indistinguibles de la población general7.
Cabe destacar que las pacientes con ST tienen mayor riesgo de mortalidad que la población general, y su expectativa de vida se reduciría en aproximadamente 13 años, principalmente por enfermedad CV8.
Para la escritura del presente manuscrito han sido revisadas las bases de datos PubMed, UpToDate e ISI web of Knowledge, utilizando la palabra clave: Síndrome de Turner. Los criterios de inclusión fueron artículos relacionados con el tema y la selección se basó en títulos y/o abstracts, disponibilidad de artículos completos y publicaciones en español o inglés. Los que no cumplieron los criterios mencionados fueron excluidos. Se encontraron 102 artículos. De éstos, 13 fueron seleccionados y 89 excluidos. Las referencias de los artículos incluidos también fueron evaluadas para identificar estudios relevantes no detectados por la búsqueda electrónica.
Bases genéticas del fenotipo
El diagnóstico se confirma mediante el análisis del cariotipo, a través de la identificación de la constitución de los cromosomas. La etiología del ST fue dilucidada en 1959, cuando C.E. Ford describió su base genética9. La ausencia parcial o total del cromosoma X se produce por una incompleta disyunción en la gametogénesis10.
Un 50% de los casos de ST corresponde a una monosomía del cromosoma X (45X), que manifiesta el fenotipo más alterado11. Un 30% corresponde a mosaicos 45X/46XX, de fenotipo más leve. La presencia del cromosoma Y otorga un 10% de riesgo de desarrollar gonadoblastoma8, por lo que debe ser investigado usando métodos específicos, como PCR en tiempo real; si se confirma, se recomienda la gonadectomía profiláctica6. En un 20% de los casos se presentan ambos cromosomas X, pero uno de ellos está incompleto o alterado: bien en forma de isocromosomas, cromosoma X en anillo o deleciones11.
Se excluyen del diagnóstico de ST individuos 45X/46XY con fenotipo masculino y mujeres con deleciones distales del brazo corto del cromosoma X, donde reside el gen SHOX (Short Stature Homeobox-containing gene on the X chromosome), las cuales pueden presentar manifestaciones esqueléticas y talla baja, pero no alteraciones CV ni infertilidad. Tampoco deben considerarse como ST deleciones distales al Xq24, las cuales producen amenorrea secundaria sin talla baja ni fenotipo característico del ST6.
La monosomía X es la que presenta mayor riesgo de malformaciones cardiacas y renales. Hasta un 40% de los mosaicos 46XX/45X pueden iniciar pubertad espontáneamente y permanecer con ovulaciones regulares durante un tiempo variable pero breve antes de desarrollar falla gonadal12. El isocromosoma Xq se asocia a enfermedades autoinmunes e hipoacusia13, mientras que las pacientes portadoras de cromosomas X en anillo son más propensas a presentar trastornos psicológicos y/o cognitivos8 (Tabla 2).
| Cariotipo | Frecuencia (%) | Fenotipo |
| 45, X | 48 | Fenotipo más severo. Alta incidencia de anomalías estructurales cardiacas y renales |
| 46, XI (Xq) | 18 | Anomalías estructurales poco comunes. Riesgo aumentado de autoinmunidad, especialmente tiroiditis y enfermedad inflamatoria intestinal, e hipoacusia |
| 45,X/46,XX | 11 | Fenotipo más leve. Mayor talla promedio. Pubertad espontánea hasta en un 40% |
| 46,Xr(X) | 10 | Reglas espontáneas en 33%. Anomalías congénitas poco comunes. Discapacidad intelectual si cromosoma en anillo. |
| 45,X/46,XY | 6 | Mayor riesgo de gonadoblastoma |
| 45,X/46,X,idic(Y) | Mayor riesgo de gonadoblastoma | |
| 46,XXp- | 1.5 | Fenotipo similar a 45,X |
| 46,XXq- | 3 | Fenotipo variable |
| OTRO | 1.5 |
La haploinsuficiencia del gen SHOX localizado en el brazo corto del cromosoma X (Xp22.3) es en gran parte responsable de la baja estaturaacientes con ST14. Entre los hallazgos fenotípicos de la deficiencia o alteración del gen SHOX se encuentran la deformidad de Madelung de la muñeca y el arqueamiento de los huesos del antebrazo y de la tibia15.
Se ha implicado tres genes que intervienen en la falla gonadal en estas pacientes: BMP15 (bone morphogenetic protein 15) ubicado en el brazo corto del cromosoma X, y los genes FMR1 y FMR2 (fragile X mental retardation), ubicados en el brazo largo5.
Respecto a los defectos cardiacos, estudios genéticos han ligado las malformaciones aórticas al Xp11.4 (y gen homólogo presente en el cromosoma Y) de las p16.
Hoy se conoce que de los embriones 45X, un 99% se aborta y sólo un 1% sobrevive. De los abortos del primer trimestre, el ST es responsable del 10% de ellos17.
El diagnóstico se efectúa en 30% de los casos antes del primer año de vida, 48% durante la niñez (1-12 años), y 22% durante la adolescencia (>12 años)18. Se ha propuesto que los sujetos 45X que sobreviven deben poseer algunas líneas celulares normales en algunos tejidos6.
Diagnóstico y screening prenatal
La ultrasonografía juega un importante rol en sospechar el ST in utero. La translucencia nucal aumentada (compartida con el Síndrome de Down y otras cromosomopatías), la presencia de higroma quístico, coartación aórtica o defectos cardíacos izquierdos, polihidroamnios y retardo del crecimiento intrauterino son característicos de ST. Independientemente de los hallazgos, el diagnóstico debe confirmarse con un cariotipo al nacer6.
El tamizaje prenatal se recomienda dada la alta frecuencia del ST y a que los tratamientos de las variadas patologías de estas pacientes podrían anticiparse. El cariotipo (gold standard actual) ofrece limitaciones importantes, tales como su costo y la demora en obtener resultados. Recientemente, se ha publicado que la PCR en tiempo real es un examen rápido y ofrece una adecuada relación costo/beneficio6.
Desarrollo cognitivo
Las pacientes con ST poseen en general inteligencia normal, aunque algunas niñas pueden presentar cierta discapacidad intelectual, especialmente las que poseen cromosoma X en anillo. En general, las pacientes con ST tienen mayor tendencia a presentar déficits en las áreas no verbales: visuo-espacial, aritmética, ejecutiva y social; y tienen mayor incidencia de trastorno por déficit atencional e hiperactividad19. En cambio, presentan un mejor desempeño promedio en lenguaje tanto expresivo como receptivo6 lo que compensaría otras deficiencias. Las últimas guías recomiendan una evaluación neuropsicológica precoz con derivación al especialista respectivo.
Dentro de las hipótesis que explican estas diferencias, se postula que la haploinsuficiencia de uno o más genes del cromosoma X podría ser la responsable. Se ha reportado que el cromosoma X materno ejercería un rol protector sobre el coeficiente intelectual global, pero la evidencia actual no es concluyente6. Desde el punto de vista neuroanatómico, se ha encontrado que las pacientes con ST presentan un volumen aberrante de sustancia gris en la corteza parieto-occipital, orbito-frontal y temporal superior en comparación con mujeres controles20.
La terapia hormonal de reemplazo (THR) puede ejercer un efecto positivo sobre algunos de estos déficits. Se ha observado que el uso de estrógenos mejora la actividad motora fina, la memoria verbal y de trabajo, componentes considerados como estrógeno-dependientes; mientras que la terapia con andrógenos como oxandrolona, puede tener un efecto positivo sobre la capacidad de atención, visualización, autopercepción y memoria21.
Defectos cardíacos
Como ya se mencionó anteriormente, las enfermedades CV son el problema de salud más serio que presentan las mujeres con ST. Su mayor morbimortalidad se debe a las malformaciones cardiacas, anomalías renales e hipertensión arterial –HTA presente en 30% y 50% de las adolescentes y adultas con ST, respectivamente–11, lo que aumenta la probabilidad de dilatación y disección aórtica5.
Dentro de las malformaciones cardiacas más comunes están la dilatación aórtica (40%), la válvula aórtica bicúspide (hasta un 30%), y la coartación aórtica (17%)22. El hallazgo de válvula aórtica bicúspide en una mujer es suficientemente específico como para derivarla para pesquisa de ST6. La disección aórtica es 6 veces más frecuente, con una incidencia de 5/100.000 adolescentes y 50/100.000 mujeres mayores de 40 años; y es la principal causa de mortalidad en todas las edades, especialmente durante el embarazo(23).
Se recomienda que todas las niñas y adolescentes con ST sean derivadas precozmente a cardiólogo para evaluación. Ésta debe incluir medición de presión arterial (PA) en manos y pies, un ECG en reposo y ecocardiograma, midiendo con este último el diámetro de la aorta ascendente (complementar en adolescentes -apenas puedan colaborar para su realizacióncon una resonancia magnética de la raíz de la aorta, para pesquisa e información adicional de anormalidades del arco aórtico y la aorta ascendente)(24) (Figura 2). Existe un parámetro denominado “ascending aorta size index” (ASI) que se utiliza en mayores de 16 años (antes de los 16 se utiliza el “TS-specific Z-score of the aorta”). Con ASI normal, en pacientes ST sin coartación aórtica, aorta bicúspide o HTA (los que implican el mayor riesgo), debería repetirse su medición cada 5 años. Si sus valores fluctúan entre 2 y 2.3 mm, estaría permitida la práctica de deportes de intensidad baja y moderada. Entre 2.3 y 2.5 mm se considera de riesgo alto, por lo que se desaconseja cualquier deporte competitivo. Sobre 2.5 mm, debe considerarse cirugía(6). Debe medirse la PA en cada control médico, ya que la HTA puede comenzar en la niñez, y a pesar de que hay evidencia limitada respecto a su uso, cada vez se está empleando más el monitoreo ambulatorio continuo de PA (Holter), que puede detectar casos de “HTA del delantal blanco” o pérdida del descenso fisiológico de PA durante la noche(23). Las guías recientes recomiendan el inicio precoz y agresivo de tratamiento antihipertensivo, con betabloqueadores y/o antagonistas del receptor de angiotensina II(6).
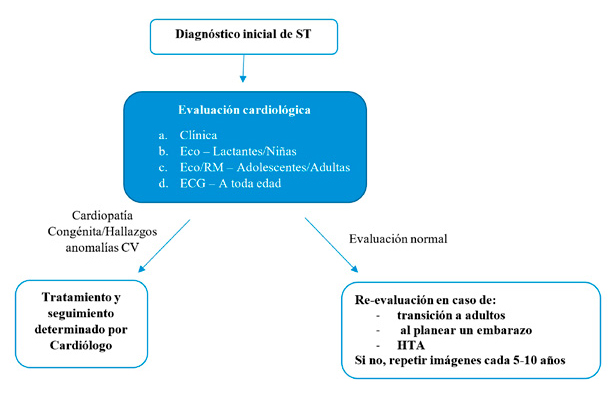
Figura 2. Evaluación cardiológica en ST. Adaptado de Pediatr Rev. 2008; 29(7): 219-227
Aspectos Otorrinolaringológicos
Las pacientes con ST presentan enfermedades del oído medio e hipoacusia con mayor frecuencia que la población general, y éstos comienzan en la primera década de la vida.
Entre un 50 y un 60% de las pacientes con ST tienen historia de otitis media aguda recurrente (OMR)(25). Se ha reportado 10-47% de hipoacusia de conducción, y ya 11% presenta hipoacusia neurosensorial (HNS) entre los 11 y 20 años, aumentado esta última su prevalencia con la edad. Los casos de hipoacusia de conducción se relacionan con los episodios de OMR, mientras que la HNS se relacionaría posiblemente con escasos niveles circulantes de estrógenos y la presencia del cromosoma X paterno(26).
Desde el punto de vista anatómico, estas pacientes presentan orejas de implantación baja, pabellones auriculares rotados, hipoplasia de los vasos linfáticos, ostium timpánico anormal, hipotonía del músculo tensor del velo del paladar y braquicefalia con paladar ojival, que provocan una horizontalización anormal de la trompa y posible disfunción palatina. En el oído interno, presentan una menor cantidad de células sensoriales cocleares al nacer, provocado una disfunción a este nivel(26). Dentro de las alteraciones palatinas, se describen crestas palatinas laterales prominentes y fisura palatina en 2% de los casos(27).
Tanto las deformidades del pabellón auricular como los episodios de OMR son más frecuentes en las pacientes portadoras de monosomía X que en pacientes con mosaicos o cromosomas X parciales con deleciones menores27. Respecto al control y seguimiento de patologías del ámbito otorrinolaringológico, se recomienda iniciar el tamizaje con Potenciales Evocados Auditivos de Tronco Cerebral al nacimiento, luego audiometrías a los 6, 12 y 36 meses. En cada control pediátrico debe realizarse otoscopía y preguntar dirigidamente por hipoacusia. Las guías actuales recomiendan realizar audiometrías cada 5 años por toda la vida6. Las otitis deben tratarse en forma precoz y agresiva. En caso de otitis media aguda recurrente u otitis con efusión persistentes (> 3 meses) debe derivarse a ORL28.
Talla baja y tratamiento
La talla baja es uno de los rasgos más constantes dentro del síndrome, afectando prácticamente al 100% de estas pacientes. Desde el nacimiento, estas pacientes ya presentan una longitud 2-3 cm inferior a la talla promedio de RN sanos. La velocidad de crecimiento (VC) se enlentece en los tres primeros años de vida y se mantiene lenta durante la niñez, sumado a la ausencia de estirón puberal provocado por la falta de hormonas sexuales5. Así, la talla final en pacientes sin terapia hormonal se estima en torno a 142 cm, aproximadamente 20 cm inferior a lo normal11. Un estudio chileno publicado el año 2002 que evaluó a 83 pacientes con ST sin tratamiento con hormona de crecimiento (HC), mostró que la longitud de nacimiento era de 46.8 cm y la altura final, fue de 138 ± 7 cm29.
La secreción de HC es habitualmente normal en estas pacientes, y sólo debiera hacerse un test de estimulación de HC en caso de que el crecimiento sea claramente anormal en relación a lo esperado, evaluado mediante las curvas de crecimiento específicas para ST24 (Figura 3). El uso de HC está aprobado tanto por la Food and Drug Administration (FDA) de EEUU como por la European Medicines Agency (EMA), y la dosis recomendada a utilizar es de 53 ug/kg/d según las guías norteamericanas27, y 45 – 50 ug/kg/d según las guías europeas6 (casi dos veces mayor que en casos de deficiencia de HC), con monitorización de niveles de IGF1 y del metabolismo glucídico30. No hay consenso respecto al momento exacto en el que debiera iniciarse el tratamiento con HC. Actualmente se recomienda que el tratamiento se inicie cuando se comprometa la VC y se confirme talla baja, entre los 4 y 6 años de edad según las guías6. Existen estudios que concluyen que iniciando el tratamiento entre los 2 y 4 años de edad se alcanzaría una talla adulta dentro de la talla objetivo familiar, sin embargo, estos estudios no llegan a talla final, sino sólo proyección de talla31. Sólo cuatro ensayos clínicos aleatorizados (ECA) han demostrado la eficacia del tratamiento con HC, con incrementos de talla entre 5 a 8 cm luego de 5-7 años de tratamiento31,32; pero sólo uno presentó resultados de talla final, con ganancia de 7,2 cm iniciando la terapia a los 9.6 ± 2.6 años33. De acuerdo a esto, una ganancia de 1 cm/año puede estimarse como adecuada6. Los factores predictores de mayor talla final adulta incluyen: talla alta relativa a inicio de la terapia, mayor talla parental, menor edad al inicio de la terapia, mayor duración de la terapia y dosis de HC24.
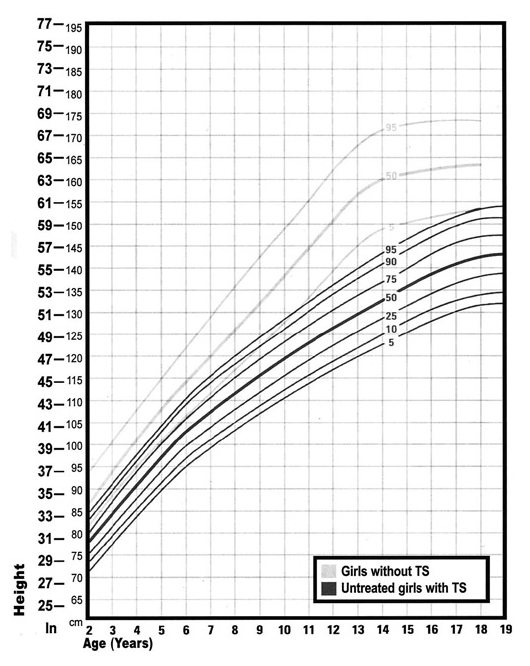 Figura 3. Curva de crecimiento para niñas con ST. (extraída con autorización de Turner Society Syndrome of the US, desde Pediatrics 2003; 111(3): 692-702.
Figura 3. Curva de crecimiento para niñas con ST. (extraída con autorización de Turner Society Syndrome of the US, desde Pediatrics 2003; 111(3): 692-702. En niñas cuyo diagnóstico ocurrió sobre los 9 años o en aquellas con talla baja extrema, se puede considerar el uso de dosis mayores de HC (hasta 68 ug/kg/día) y/o la adición de un esteroide anabólico no aromatizable como la oxandrolona. Éste debiera usarse a una dosis ≤ 0.05 mg/kg/d (dosis mayores pueden inducir virilización), monitorizando enzimas hepáticas. El tratamiento debiera continuarse hasta que no quede potencial de crecimiento (edad ósea ≥ 14 años y VC < 1 cm/año)24.
Otro factor que influye en la talla final adulta, es la edad a la que se inicie la inducción puberal. Normalmente la terapia con estrógenos se inicia a los 12 años, pero recientemente se ha demostrado que la combinación de HC con dosis muy bajas de estrógenos en la niñez podría mejorar el crecimiento y otorgar otros beneficios potenciales34 como se comentará a continuación.
Introducción precoz estrógenos
Si bien aproximadamente el 25% de las niñas con ST puede alcanzar cierto grado de desarrollo puberal24, el 90% requerirá en algún momento THR para el inicio, progresión y mantención de los signos puberales. Como en cualquier paciente con hipogonadismo, el momento, la dosis y la forma de administración de la THR debiese ser lo más parecido posible al proceso fisiológico.
El consenso internacional aún es iniciar la THR a los 11 - 12 años, si no hay pubertad espontánea y los niveles de FSH están elevados35. Como se mencionó anteriormente, algunos trabajos han demostrado que el uso de estrógenos antes de los 12 años puede tener efectos beneficiosos. En el ECA y doble ciego de Ross y cols. del año 2011, se demostró que la administración oral de etinilestradiol desde los 5 años de edad (25 ng/kg de los 5 a 8 años y 50 ng/kg de los 8 a los 12 años) asociado a HC provocaba un aumento en la talla final adulta de 0.32 DE (2.1 cm) en comparación con HC sola, además de beneficios en cognición, percepción personal, velocidad de procesamiento no verbal, desempeño motor y memoria34. Otro efecto beneficioso del uso de dosis muy bajas de estrógenos durante la niñez fue publicado por Quigley y cols. 3 años después, demostrando en forma significativa un inicio más precoz de la telarquia, mayor tamaño uterino y un tiempo de pubertad más prolongado –más similar al proceso fisiológico– en las niñas que iniciaron etinilestradiol a los 5 años, comparadas con el grupo control36. A pesar de ello, las guías 2017 sugieren no usar en forma rutinaria dosis bajas de E2 en niñas prepuberales dado la falta de evidencia respecto a la seguridad a largo plazo6.
La inducción de la pubertad se inicia mediante la administración de estrógenos –el fármaco recomendado en la guía del Grupo de Estudio sobre Síndrome de Turner24 es el 17 β estradiol– a dosis bajas (1/8 o 1/10 de la dosis de adulto) con aumentos progresivos por un periodo de 2 - 4 años hasta alcanzar la dosis de adulto. El año 2011 Menéndez y cols. publicaron un artículo con los esquemas más frecuentemente usados y los tipos de estrógenos disponibles en Chile37. El fármaco más usado en nuestro país es el 17β estradiol en presentación oral (Primaquin), sin embargo, cada vez más se está implementado el uso de parches transdérmicos, ya que ofrecen un método de administración más fisiológico que el oral al eliminar el primer paso hepático. Existe evidencia contradictoria respecto a los beneficios en densidad mineral ósea(35,38), pero sí se ha observado que el uso de estrógenos transdérmicos (ETD) logra alcanzar un mayor tamaño uterino. Por otro lado, los estrógenos orales tienen un impacto más favorable en colesterol HDL y LDL, sin reportar diferencias en los niveles de triglicéridos o insulina39. En mujeres postmenopáusicas los estrógenos orales estarían asociados a un mayor riesgo tromboembólico35.
El problema radica en que en nuestro país no existen presentaciones de E2 para vía transdérmica (parches o gel) que permitan iniciar fácilmente las dosis recomendadas para inducción puberal. Habría que dividir los parches que entregan 50 ug diarios de E2 en 8 para obtener la dosis indicada, sin embargo, éstos no están confeccionados para ser cortados37. No obstante, existen publicaciones en las cuales se ha probado la efectividad del corte de los parches, midiendo los niveles séricos de E2 alcanzados y el resultado clínico en estadíos de Tanner mamario40.
Fertilidad y embarazo
Las guías 2017 recomiendan informar a las pacientes con ST que su probabilidad de concebir espontáneamente (si existiese), disminuye rápidamente con la edad y debería conversarse sobre las terapias de fertilidad a una edad temprana6. El embarazo en ST implica tanto riesgos fetales como maternos. Existe 3.5 veces mayor riesgo de aborto que la población general. De un estudio de 160 embarazos en ST, hubo un 58% de hijos vivos, y de éstos, 34% presentó cromosomopatías, principalmente ST o Síndrome de Down41. Otro estudio retrospectivo de 115 embarazos en pacientes con ST reportó mayor incidencia de preeclampsia (6.3 vs 3% en población general), de cesáreas (35% vs 11.8%) y menor edad gestacional y peso de nacimiento42.
Las guías actuales establecen que la opción más segura para el binomio madre-hijo (a) es la adopción, y mencionan un menor riesgo fetal con la ovodonación6.
Respecto a la criopreservación ovárica, es una opción en pacientes ST con función ovárica persistente (con o sin pubertad). Los elementos que otorgan el mejor pronóstico para obtener ovocitos son FSH < 11 mUI/ml y AMH > 2 pmol/L43. Se recomienda realizarla en adolescentes mayores de 12 años. Lamentablemente, hasta nuestro conocimiento, no se han descrito niños nacidos mediante esta técnica. La transición de las adolescentes con ST hacia el cuidado adulto es trascendental y merece un capítulo aparte.
Conclusiones
Este artículo da cuenta de avances en comprender y tratar los principales problemas que presentan niñas y adolescentes con ST. Hemos mejorado, pero aún queda mucho por hacer en beneficio de la salud integral de las pacientes con ST, tanto en aspectos de crecimiento físico, salud psicológica, anticipación de problemas CV y otorrinos, y recomendaciones de fertilidad. Considerando que en nuestro país hay en nuestro país, según entendimiento de los autores, sólo 11 centros, sólo 11 centros que cuentan con un equipo multidisciplinario capacitado para atender a pacientes con ST, sería recomendable iniciar un registro nacional único de ST, ya que en Chile no contamos con un registro de este tipo, y la experiencia adquirida, así como las posibilidades de investigación está limitada al pequeño número de casos en cada institución.
Referencias
- Castelo-Branco C. Management of Turner syndrome in adult life and beyond. Maturitas 2014; 79(4): 471-475.
- Ullrich O. Über typische Kombinationsbilder multipler Abartungen. Eur J Pediatr 1930; 49: 271-276.
- Classic pages in obstetrics and gynecology by Henry H. Turner. A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endocrinology 1938; 23: 566-574. Am J Obstet Gynecol 1972; 113: 279.
- Levitsky L, O’Donnell A, Hayes F, Lin A. Turner syndrome: update on biology and management across the life span. Curr Opin Endocrinol Diabetes Obes 2015, 22: 65-72.
- Backeljauw P. Clinical manifestations and diagnosis of Turner syndrome. En: UpToDate, Martin K, Hoppin A (Ed), UpToDate, 2016.
- Gravholt C, Andersen N, Conway G, Dekkers O, Geffner M, Klein K, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol 2017; 177(3): G1-G70.
- Carvalho AB, Guerra-Junior G, Baptista MT, Marques-de-Faria AP, Lemos- Marini SH, Maciel-Guerra AT. Turner syndrome: a pediatric diagnosis frequently made by non-pediatricians. J Pediatr (Rio J). 2010; 86: 121-125.
- Elsheikh M, Dunger, Conway G, Wass H. Turner’s Syndrome in Adulthood. Endoc Rev 2002; 23(1): 120-140.
- Ford CE, Jones KW, Polani PE, De Almeida JC, Briggs JH. A sex-chromosome anomaly in a case of gonadal dysgenesis (Turner’s syndrome). Lancet 1959; 4(1): 711-713.
- Collin J. An Introduction to Turner Syndrome. Paediatric Nursing 2006; 18(10): 38-43.
- Barreda AC, González I, Gracia R. Síndrome de Turner. Protoc Diagn Ter Pediatr 2011; 1: 218-227.
- Pasquino AM, Passeri F, Pucarelli I, Segni M, Municchi G. Spontaneous pubertal development in Turner’s syndrome. Italian Study Group for Turner’s Syndrome. J Clin Endocrinol Metab 1997; 82: 1810-1813.
- Stratakis CA, Rennert OM. Turner syndrome: molecular and cytogenetics, dysmorphology, endocrine, and other clinical manifestations and their management. Endocrinologist 1994; 4: 442-453.
- del Rey G. El gen SHOX y el crecimiento corporal: descripción, estructura y nuevas técnicas de diagnóstico. BAG, J Basic Appl Genet 2010; 21(2): 1-9.
- Child C.J, Kalifa G, Jones C, Ross J, Rappold G, Quigley C, et al. Radiological Features in Patients with Short Stature Homeobox-Containing (SHOX) Gene Deficiency and Turner Syndrome before and after 2 Years of GH Treatment. Horm Res Paediatr 2015; 84 (1): 14-25.
- Bondy C, Bakalov VK, Cheng C, Olivieri L, Rosing DR, Arai AE. Bicuspid aortic valve and aortic coarctation are linked to deletion of the X chromosome short arm in Turner syndrome. J Med Genet 2013; 50: 662.
- Morgan T. Turner Syndrome: Diagnosis and Management. Am Fam Physician 2007; 76(3): 403-410.
- 18. Massa G, Verlinde F, De Schepper J, Thomas M, Bourguignon J.P, Craen M, et al. Trends in age at diagnosis of Turner syndrome. Arch Dis Child 2005; 90: 267-268.
- Loscalzo M. Turner Syndrome. Pediatr Rev 2008; 29(7): 219-227.
- Hong D, Hoeft F, Marzelli M, Lepage J, Roeltgen D, Ross J, et al. Influence of the X-Chromosome on Neuroanatomy: Evidence from Turner and Klinefelter Syndromes. J. Neurosci 2014; 34(10): 3509-3516.
- Nehama Zuckerman-Levin N, Frlova-Bishara T, Militianu D, Levin M, Aharon-Peretz J, Hochberg Z. Androgen Replacement Therapy in Turner Syndrome: A Pilot Study. J Clin Endocrinol Metab 2009; 94(12): 4820- 4827.
- Mortensen KH, Andersen NH, Gravholt CH. Cardiovascular phenotype in Turner syndrome-integrating cardiology, genetics, and endocrinology. Endocr Rev 2012; 33(5): 677-714.
- Turtle E.J, Sule A.A, Webb D.J, Bath L.E. Aortic dissection in children and adolescents with Turner syndrome: risk factors and management recommendations. Arch Dis Child 2015; 100: 662-666.
- Bondy C.A, Turner syndrome study group. Care of Girls and Women with Turner Syndrome: A Guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab 2007; 92(1): 10-25.
- Chan K, Wang P, Lo F. Otologic and Audiologic Features of Ethnic Chinese Patients with Turner Syndrome in Taiwan. J Formos Med Assoc 2012; 111(2): 94-100.
- Alves C, Oliveira CS. Hearing loss among with Turner’s syndrome: literature review. Braz J Otorhinolaryngol 2014; 80: 257-263.
- Makishima T, King K, Brewer C, Zalewski C, Butman J, Bakalov V. Otolaryngologic markers for the early diagnosis of Turner syndrome. Int J Pediatr Otorhinolaryngol 2009; 73(11): 1564-1567.
- Frías J, Davenport M. Health Supervision for Children with Turner Syndrome. Pediatrics 2003; 111(3): 692-702.
- Román R, Vallejos ME, Muñoz M, Schneider R, Youlton R, Henriquez C, et al. Síndrome de Turner: Crecimiento y descripción clínica en 83 niñas chilenas. Rev Méd Chile 2002; 130(9): 977-984.
- Davenport ML. Approach to the patient with Turner syndrome. J Clin Endocrinol Metab 2010; 95: 1487-1495.
- Baxter L, Bryant J, Cave CB, Milne R. Recombinant growth hormone for children and adolescents with Turner syndrome. Cochrane Database Syst Rev 2007; (1): CD003887.
- Sánchez M, Muñoz A, Ferrer M, Labarta JI, Garagorri JM. Hormona de crecimiento y síndrome de Turner. An Pediatr (Barc) 2017; 86(2): 81-86.
- Stephure DK. Canadian Growth Hormone Advisory Committee. Impact of growth hormone supplementation on adult height in turner syndrome: results of the Canadian randomized controlled trial. J Clin Endocrinol Metab 2005; 90: 3360-3366.
- Ross J, Quigley C, Cao D, Feuillan P, Kowal K, Chipman J, et al.Growth Hormone plus Childhood Low-Dose Estrogen in Turner’s Syndrome. N Engl J Med 2011; 364: 1230-1242.
- Gawlik A, Hankus M, Such K, Drosdzol-Cop A, Madej P, Borkowska M, et al. Hypogonadism and Sex Steroid Replacement Therapy in Girls with Turner Syndrome. J Pediatr Adolesc Gynecol 2016; 29(6): 542-550.
- Quigley C, Wan X, Garg S, Kowal K, Cutler G, Ross J. Effects of Low-Dose Estrogen Replacement During Childhood on Pubertal Development and Gonadotropin Concentrations in Patients With Turner Syndrome: Results of a Randomized, Double-Blind, Placebo-Controlled Clinical Trial. J Clin Endocrinol Metab 2014; 99(9): E1754-1764.
- Menéndez M, Rumie K, García H. Inducción de la pubertad en el síndrome de Turner. Rev Chil Pediatr 2011; 82 (5): 433-439.
- Cintron D, Rodriguez-Gutierrez R, Serrano V, Latortue-Albino P, Erwin P, Murad M. Effect of estrogen replacement therapy on bone and cardiovascular outcomes in women with turner syndrome: A systematic review and metaanalysis. Endocrine 2017; 55(2): 366-375.
- Zaiem F, Alahdab F, Nofal A, Murad M, Javed A. Oral versus transdermal estrogen in turner syndrome: A systematic review and meta-analysis. Endocr Pract. 2017 Jan 17. [Epub ahead of print]
- Ankarberg-Lindgren C, Kriström B, Norjavaara E. Physiological estrogen replacement therapy for puberty induction in girls. Horm Res Paediatr 2014; 81: 239-244.
- Tarani L, Lampariello S, Raguso G, Colloridi F, Pucarelli I, Pasquino AM, et al. Pregnancy in patients with Turner’s syndrome: six new cases and review of literature. Gynecological Endocrinology. 1998 Apr; 12(2): 83-87.
- Oktay K, Bedoschi G, Berkowitz K, Bronson R, Kashani B, McGovern P, et al. Fertility Preservation in Women with Turner Syndrome: A Comprehensive Review and Practical Guidelines. J Pediatr Adolesc Gynecol. 2016; 29(5):409-416.
- Borgström B, Hreinsson J, Rasmussen C, Sheikhi M, Fried G, Keros V, et al. Fertility preservation in girls with turner syndrome: prognostic signs of the presence of ovarian follicles. J Clin Endocrinol Metab. 2009; 94(1):74-80.