Histiocitosis de células de Langerhans con afectación hipofisaria, seguimiento endocrinológico en paciente adulto: A propósito de un caso
Bryan Acosta1*, Florencia Dorfman1, María. M. Piñeyro1.
Langerhans Cell Histiocytosis with Pituitary Involvement, Endocrinological Follow-up in an Adult Patient: A Case Report
- Clínica de Endocrinología y Metabolismo, Hospital de Clínicas, Facultad de Medicina, Universidad de la República, Montevideo, Uruguay.partamento de Nutrición. Facultad de Medicina Norte. Universidad de Chile. Santiago, Chile.
*Correspondencia:Bryan Acosta / abaaz_8j@hotmail.com
Recibido: 02-08-2024
Aceptado: 10-09-2024
Resumen: La histiocitosis de células de Langerhans (HCL) es una rara afección
histiocítica que se distingue por el crecimiento excesivo de células del
sistema fagocítico mononuclear (macrófagos, células dendríticas, monocitos)
en diversos órganos y sistemas. La afectación de la glándula hipófiisis se
reporta en 5-50% de los pacientes con HCL, lo más frecuente la deficiencia
de arginina vasopresina en 50% de los casos. Una minoria también presenta
disfunción de hormonas de la adenohipófisis. Presentamos el caso de
un paciente masculino de 33 años, diagnosticado de HCL en contexto de
infecciones óticas a repetición, con tratamiento quimioterápico hasta en 3
ocasiones por recidiva tumoral. En el seguimiento se evidencia deficiencia
de arginina-vasopresina. Asimismo, se diagnostica panhipopituitarismo. Nos
enfocaremos en la definición, fisiopatología, epidemiología, y cómo realizar el
seguimiento endocrinológico en pacientes adultos con HCL. La afectacion de
la hipófisis puede resultar un desafío diagnóstico. Es fundamental reconocer
esta manifestacion clínica para un diagnóstico y tratamiento adecuado, ya
que sin tratamiento el paciente puede enfrentar un riesgo vital.
Palabras clave: Deficiencia arginina-vasopresina; Histiocitosis de células de
Langerhans; Histiocitosis X; Panhipopituitarismo.
Abstract: Langerhans cell histiocytosis (LCH) is a rare histiocytic disorder
characterized by the excessive proliferation of cells from the mononuclear
phagocyte system (macrophages, dendritic cells, monocytes) in various organs
and systems. Pituitary gland involvement is reported in 5-50% of patients with
LCH, with the most common issue being arginine vasopressin deficiency in
50% of cases. A minority also present with adenohypophysis hormone dysfunction.
We present the case of a 33-year-old male patient diagnosed with
Langerhans cell histiocytosis (LCH) in the context of recurrent ear infections,
with chemotherapy treatment administered up to three times due to tumor
relapse. Follow-up reveals arginine-vasopressin deficiency. Additionally, the
patient is diagnosed with panhypopituitarism.
We will focus on the definition, pathophysiology, epidemiology, and how
to conduct endocrinological follow-up in adult patients with LCH. Pituitary
gland involvement can pose a diagnostic challenge. Recognizing this clinical
manifestation is crucial for accurate diagnosis and appropriate treatment, as untreated patients may face life-threatening risks.
Keywords: Arginine-vasopressin deficiency; Histiocytosis X; Langerhans cell
histiocytosis; Panhypopituitarism.
Introducción
La histiocitosis de células de Langerhans (HCL), asi llamada por la infiltración tisular de células con estructura y marcadores específicos de las células de Langerhans (CD1a+/CD207+), presentes en las lesiones entre un 1% a > 70% (promedio de 8%)1. En 1953, fue agrupada bajo el término histiocitosis X debido al origen incierto de las células. En los años 80, gracias a la microscopía electrónica, se pudo diferenciar los tipos de histiocitosis, lo que llevó a la definición de HCL por parte de la sociedad de histiocitosis2.
Se caracteriza por activación constitutiva de la vía MAPK, lo que produce una multiplicación clonal de precursores mieloides que se diferencian en CD1a+/CD207+ en los tejidos afectados. Estas células tienen una baja tasa de proliferacion pero son menos sensibles a la apoptosis, lo que resulta en lesiones persistentes. Ocurre de manera esporádica3.
La histiocitosis de células de Langerhans (HCL) puede presentarse de varias maneras: unifocalmente, con una lesión única en un solo órgano; multifocalmente en un solo sistema, con dos o más lesiones que afectan un solo órgano; y de forma multisistémica, con dos o más órganos comprometidos4. Se estima una prevalencia entre 1 y 1,5 casos por millón de habitantes/año en población pediátrica2,5. La epidemiología es desconocida en adultos.
El diagnóstico se realiza mediante la histopatología del tejido afecto, junto con inmunohistoquímica que evidencie positividad del marcador CD1a2. Las pautas actuales proponen que tanto para la estadificación inicial como el seguimiento se realicen con tomografía por emisión de positrones con flúor‐18‐fluorodeoxiglucosa (18-PET- FDG)4,5.
Esta patología puede comprometer diversos sistemas, que incluyen el sistema esquelético, pulmonar, timo, sistema hepatobiliar, sistema digestivo, sistema nervioso central (SNC), los tejidos blandos de la cabeza y el cuello, glándulas salivales y raramente tiroides6. Las manifestaciones cutáneas y esqueléticas son las más comunes en pacientes adultos, es especial de cráneo y vertebras.
En lo endocrinológico, la afectación de la glándula hipófiisis se reporta en 5-50% de los pacientes con HCL, lo mas frecuente la deficiencia de arginina vasopresina en 50% de los casos7. Una minoría (5-20%) también presenta disfunción de hormonas de la adenohipófisis4,8,9. En la resonancia magnética nuclear (RMN) se puede encontrar a nivel de sistema nervioso central las siguientes manifestaciones: pérdida del punto brillante de la glándula pituitaria, engrosamiento del tallo pituitario >3 mm con realce, lesión masiva con isointensidad y realce homogéneo bajo imágenes potenciadas en T16.
Reportamos el caso de un paciente con HCL diagnosticado en la pubertad que en la evolución de la enfermedad agrega múltiples déficits hormonales. Representa un desafío dada la poca frecuencia de la patología.
Caso clínico
Paciente de sexo masculino, de 33 años. Diagnosticado de HCL a los 13 años de edad, en contexto de infecciones óticas recurrentes y pobre respuesta a los tratamientos tradicionales. Se realiza biopsia del sitio de lesión donde se evidencia HCL. Recibe tratamiento quimioterápico en 3 ocasiones con: Vinblastina + Prednisona / Citarabina + Prednisona / 2 -Clorodeoxiadenosina.
Al inicio de su tercera quimioterapia, el paciente presenta polidipsia de predominio nocturno con avidez por agua fría, llega a consumir 15 litros de agua por día, acompañado de poliuria. Se realiza prueba de restriccion hidrica que fue positiva, que muestra hipernatremia con osmolaridad urinaria baja (<300 mOSM).
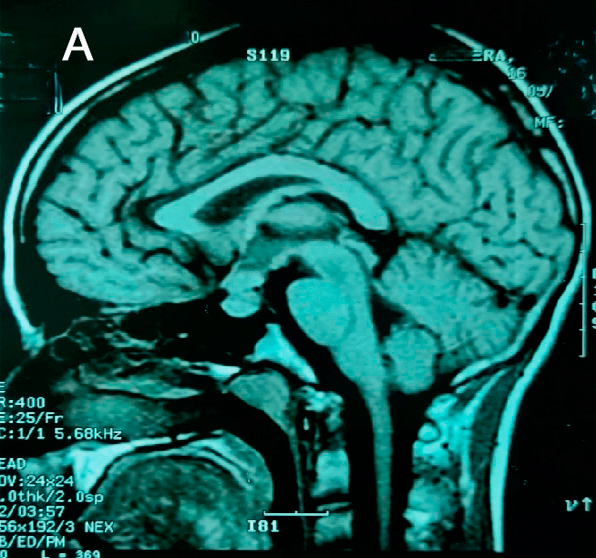
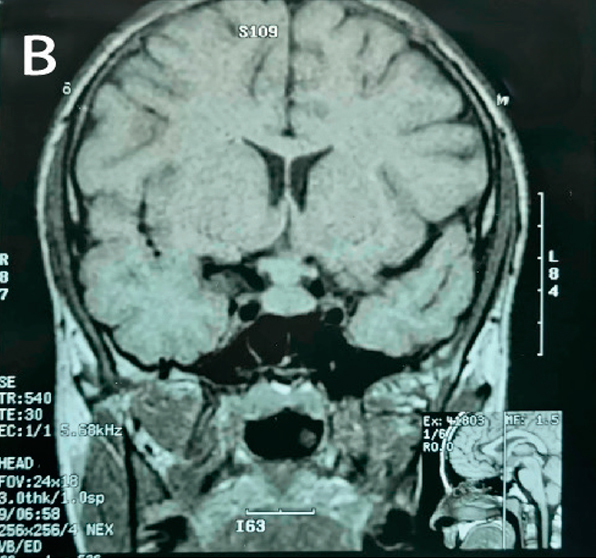
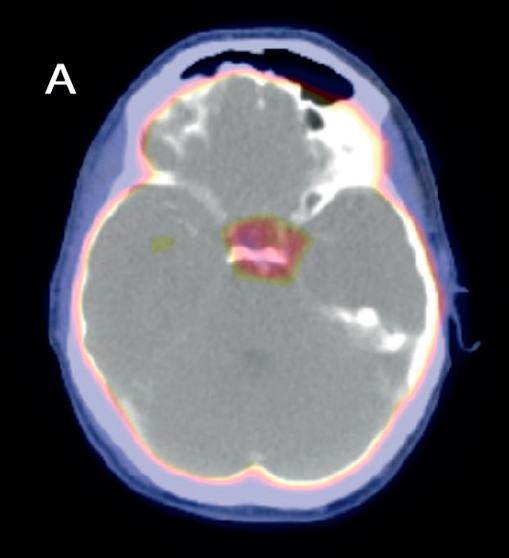
Se realiza diagnóstico de deficiencia de arginina-vasopresina central y se inicia desmopresina intranasal a demanda. Se realiza una RMN con enfoque selar, que reporta una lesión hipofisaria que compromete glándula y tallo hipofisario (Figura 1).
Se realiza estudio de las hormonas de la adenohipofisis. Se realiza diagnóstico de hipogonadismo hipogonadotrófico con los siguientes valores: testosterona 1,5 mUI/mL (VR: 2,49-8-26), hormona luteinizante <0,1 mUI/mL (VR: 0,6-12,1), hormona folículo estimulante 0,1 mUI/ mL (VR: 1-12). Se inicia sustitución con enantato de testosterona 250 mg /cada 28 días, previo hematocrito y enzimas hepáticas, los cuales resultaron normales. Asimismo, se diagnostica hipotiroidismo secundario con los siguientes valores: TSH 0,43 ng/mL (VR: 0,23-4,7), T4L 0,34 ng/mL (VR: 0.93-1.70) por lo que se inicia tratamiento con levotiroxina 100 ug/día. Ademas, se evidencia una insuficiencia suprarrenal secundaria con un cortisol AM de 3 ug/dL (VR: 5-25), y se inicia tratamiento con hidrocortisona 10 mg (6 am)/5mg (5 pm). El paciente presenta deficiencia de argininavasopresina, inicialmente manejada con desmopresina a demanda. Estuvo hospitalizado en 3 ocasiones por intoxicación hídrica. Actuamente, se encuentra estable con desmopresina reglada 1 puff cada 6 horas.
Se plantea afectacion del tallo hipofisario y de la glándula hipófisis por la HCL, basado en el diagnóstico de HCL obtenido mediante biopsia de lestion en la mastoides. No se consideró necesario realizar biopsia de la lesion hipofisaria. El paciente continua con la quimioterapia, que recibe 6 ciclos, cada 3 semanas.
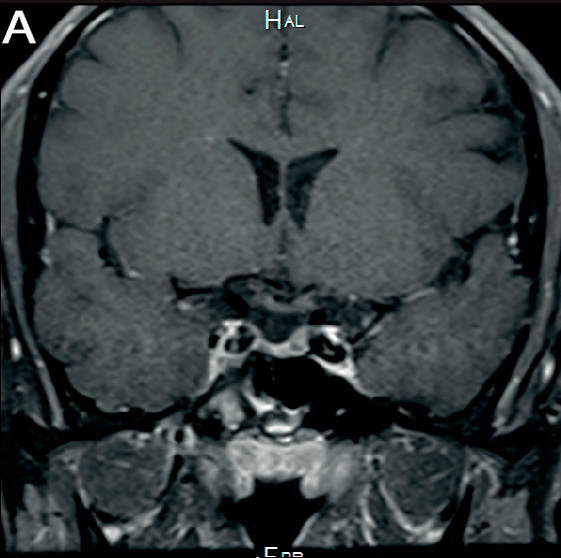
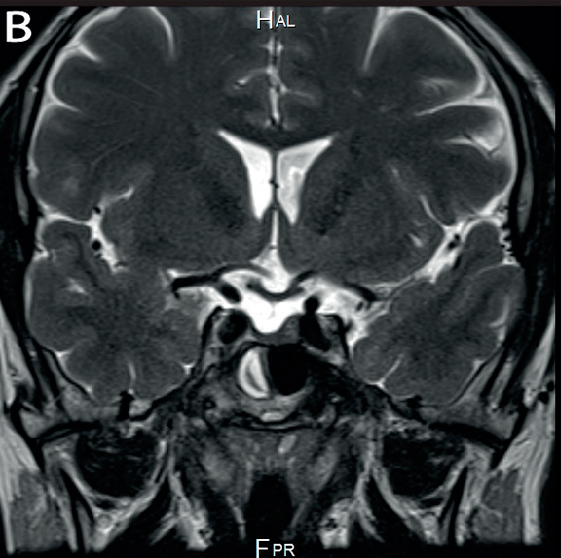
En la evolución se realiza RNM de control 2 años posteriores a primera RMN, que evidencia disminución del tamaño de la lesión tanto en el tallo hipofisario como de la hipófisis (Figura 2).
Actualmente, el paciente se encuent ra con sustitución en los 3 ejes hormonales (gonadal, tiroideo, corticosuprarrenal), con dosificaciones hormonales dentro de rango de normalidad para los ejes gonadal y tiroideo. Eje corticoideo se encuentra controlado sin elementos de síndrome de Cushing, con presión arterial normal y sin hipotensión ortostática. Asimismo, está educado para el incremento de dosis ante situaciones de estrés y lleva chapa identificatoria. Está en tratamiento para deficiencia de arginina-vasopresina, sin presentar poliuirodipsia y con electrolitos séricos normales. Además, presenta un campo visual por confrontación normal.
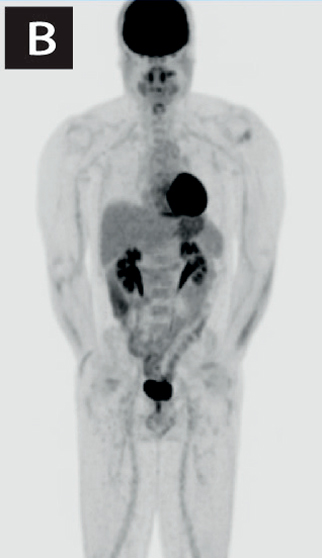
En el seguimiento, se solicitó una tomografía por emisión de positrones (PET-CT) en búsqueda de lesiones en otras regiones corporales. Se evidenció un aumento de captación en sector lateral izquierdo en región selar sin correlato morfológico, lo que es inespecifico. No se evidencian lesiones en pulmones ni en estructuras óseas (Figura 3). Se plantea que el paciente está en remisión, y se continuará su seguimiento.
Discusión
La HCL es una enfermedad relativamente rara caracterizada por la proliferación clonal anormal y la acumulación de células dendríticas presentadoras de antígenos en múltiples tejidos y órganos, con una amplia variedad de manifestaciones clínicas y presentaciones histológicas. Las células dendríticas que se encuentran en las lesiones de HCL son precursoras que se originan en la médula ósea, migran a los sitios de lesión y se diferencian en células positivas para langerina (CD207). La HCL está asociada con una significativa morbilidad. Puede afectar varios sistemas, incluida la afectación a nivel hipofisario que puede conducir a hipopituitarismo con necesidad de reemplazo hormonal de los ejes hormonales, como en este caso. La deficiencia de arginina-vasopresina es la manifestación endócrina mas frecuente. Se puede presentar incluso años antes de presentar el resto de compromiso a diferentes órganos/sistemas1.
La supervivencia para paciente con afectación unifocal es excelente, mientras que las tasas de mortalidad pueden alcanzar hasta un 20% en aquellos con disfunción multi-sistema4.
Cuando se presente deficiencia de arginina-vasopresina, es crucial realizar RMN con enfoque selar para valorar posibles lesiones hipofisarias. Ademas, se debe llevar a cabo una evaluación de los ejes hormonales hipofisarios (corticosuprarrenal, tiroideo, gonadal, somático, lactotropo). En caso de su afectación se debe iniciar la correspondeinte terapia de reemplazo hormonal. Es recomendable evaluar los ejes hormonales incluso en ausencia de una imagen clara de lesión hipofisaria, ya que estas disfunciones pueden estar presentar sin lesiones evidentes10.
Dentro de los factores de riesgo para desarrollar HCL se incluyen ser de origen latino, antecedentes de infección tracto urinario materno durante la gestación, transfusiones sanguíneas en la infancia, hacinamiento, bajo nivel escolar, infecciones neonatales, exposición a solventes, antecedentes familiares de enfermedad tiroidea y fertilización in vitro. Por otro lado, factores protectores incluyen pertenecer a la raza negra, recibir vacunación infantil y tomar suplementos vitamínicos11.
En pacientes adultos, el seguimiento debe ser de por vida dado que los déficits hormonales pueden manifestarse incluso años después del diagnóstico inicial. Entre las hormonas hipofisarias, la hormona del crecimiento es la más afectada. Hasta ahora, no se ha encontrado una forma de prevenir de secuelas a largo plazo hipotalámico-hipofisarias. Se han identificado 3 factores de riesgo para el desarrollo de deficiencia de arginina-vasopresina: enfermedad multisistémica, enfermedad activa o reactivada por tiempos prolongados, y presencia de lesiones óseas en arcas como el hueso frontal, órbitas, oído medio y mastoides, como en nuestro paciente. Ademas, se han observado 3 características que aumentan la probabilidad de desarrollo de deficiencias hormonales hipofisarias: mayor edad al diagnóstico, afectación multi-sistemica, y pocos episodios de reactivación de la enfermedad12,13. En el caso especifico de nuestro paciente, este presentó una edad mayor al promedio de inicio de la enfermedad.
Al realizar el tratamiento quimioterápico en estos pacientes se debe también iniciar sustitución hormonal, si así lo requirieran, independientemente del tipo de quimioterapia usada14.
A largo plazo se ha reportado supervivencia en HCL multisistema de un 90% en niños, en adultos no contamos con estadística. Cuando presentan recaídas de la enfermedad, estas son menos graves que la enfermedad inicial en extensión, gravedad, y mortalidad15.
En cuanto a las manifestaciones endocrinológicas, la deficiencia de arginina-vasopresina es la más frecuente. Esta manifestación debe sospecharse ante pacientes con síndrome poliurodípsico, a veces con avidez por agua fría en las noches. El diagnóstico se realiza si existe hipernatremia con hiperosmolaridad sanguínea e hipoosmolaridad urinaria. En los casos con mecanismo de la sed intacto, donde no se desarrolla hipernatremia, se puede confirmar con la prueba de restricción hídrica. En la RMN, se puede observar una pérdida de brillo de la neurohipófisis en secuencia T1 y engrosamiento del tallo pituitario2.
No existe un marcador pronóstico para la evolución hacia un daño endócrino, ya que las afectaciones de la adenohipófisis pueden presentarse con o sin déficit de arginina-vasopresina. Por lo tanto, en pacientes con HCL sin déficits hormonales, es necesario realizar un seguimiento clínico y paraclínico regular. Se recomienda buscar deficiencia de GH, con la medición de IGF-1 y eventualment test de estímulo en adultos, dado que su reemplazo mejora la sensación de bienestar y el perfil metabólico del paciente. Ademas, se debe investigar la presencia de hipogonadismo hipogonadotrópico con medición de LH-FSH y testosterona en hombres, y estradiol en mujeres. Los niveles de prolactina pueden estar elevados por efecto de masa.Se recomienda despistar déficit de ACTH con un cortisol y ACTH hora 8 am, así como hipotiroidismo secundario mediante la valoración de TSH y T4L2,4.
El tratamiento puede reducir el tamaño de las lesiones, pero en general, los ejes hormonales afectados no se recuperan. Además, puede presentarse sintomatología debido al crecimiento tumoral, como alteraciones visuales; por ello debe realizarse un examen por confrontación y campo visual computarizado2. Es importante recordar que existen otras afectaciones cerebrales no endocrinas, como cambios en el comportamiento social, memoria, apetito, saciedad y sueño4.
Si la presentación de HCL es únicamente con compromiso hipofisario, se debe hacer reemplazo hormonal correspondiente. En estos casos, el papel de la terapia sistémica no está claro. Sin embargo, la terapia sistémica se recomienda en casos con aparición reciente o lesión radiológica presente. Al inicio se recomienda evaluación de ejes hormonales hipofisarios a los 3 meses del diagnóstico de HCL y de ahí de manera anual4.
Se ha reportado que la mayoría de paciente con HCL sufren un retraso en el diagnóstico, debido a los síntomas inespecíficos de la enfermedad, lo que incrementa la morbilidad y mortalidad, como por ejemplo la falta de sustitución de ejes hormonales vitales16.
El tratamiento debe basarse a la presentación clínica del paciente, y puede incluir quimioterapia, inmunosupresión, radioterapia y sustitución hormonal. El seguimiento debe de ser multidisciplinario. Deben de estar involucrados oncología debido a su mayor prevalencia de ciertos tipos de neoplasias como leucemias, trastornos mieloproliferativos, y linfomas17 así como dermatología, neumología, hematología18 y endocrinología dadas las manifestaciones en diferentes sistemas18.
Conclusiónes
El manejo endocrinológico del paciente con HCL adulto plantea desafíos significativos, dado que el compromiso de ejes hipofisarios puede pasar desapercibido, lo que podria tener consecuencias criticas para el paciente con compromiso vital. El seguimiento a largo plazo de estos pacientes es esencial, debido al riesgo de recurrencia de la enfermedad subyacente y su eventual compromiso multisistémico. Se destaca la importancia del manejo multidisciplinario integral en pacientes portadores de HCL.
Referencias
- Allen CE, Merad M, McClain KL. Langerhans-Cell Histiocytosis. New England Journal of Medicine. 2018; 379(9): 856-868.
- de Menthon M, Meignin V, Mahr A, Tazi A. Histiocytose à cellules de Langerhans de l’adulte. Presse Med. 2017; 46(1): 55-69.
- Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood. 2020; 135(16): 1319-1331.
- Goyal G, Tazi A, Go RS, Rech KL, Picarsic JL, Vassallo R, et al. International expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults. Blood. 2022; 139(17): 2601-2621.
- Gorria F, García V, Hackenbruch E, Hermosilla S, Ipharraguerre M, Romero R. Valor del PET/CT con 18F-FDG en pacientes pediátricos con Histiocitosis de Células de Langerhans (HCL) [Monografía]. [Montevideo]: UDELAR; 2017.
- Cheng YF, Wang CC, Tsai PS, Lin DC, Huang WH. Langerhans cell histiocytosis of the thyroid mimicking thyroiditis in a boy: A case report and literature review. BMC Pediatr. 2024; 24(1): 66.
- Broadbent V, Dunger DB, Yeomans E, Kendall B. Anterior pituitary function and computed tomography/magnetic resonance imaging in patients with langerhans cell histiocytosis and diabetes insipidus. Med Pediatr Oncol. 1993; 21(9): 649-654.
- Willis B, Ablin A, Weinberg V, Zoger S, Wara WM, Matthay KK. Disease course and late sequelae of Langerhans’ cell histiocytosis: 25-year experience at the University of California, San Francisco. Journal of Clinical Oncology. 1996; 14(7): 2073-2082.
- Doleschal B, Popper U. Sustained remission of adult Langerhans histiocytosis utilizing molecular therapy. memo - Magazine of European Medical Oncology. 2021; 14(1): 115-118.
- Gulati N, Allen CE. Langerhans cell histiocytosis: Version 2021. Hematol Oncol. 2021; 39(S1): 15-23.
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children. J Am Acad Dermatol. 2018; 78(6): 1035-1044.
- Vaiani E, Malossetti C, Vega LM, Zubizarreta P, Braier J, Belgorosky A. Predictor Variables of Developing Anterior Pituitary Deficiencies in a Group of Paediatric Patients with Central Diabetes Insipidus and Langerhans Cell Histiocytosis. Horm Res Paediatr. 2017; 87(1): 51-57.
- Vaiani E, Felizzia G, Lubieniecki F, Braier J, Belgorosky A. Paediatric Langerhans Cell Histiocytosis Disease: Long-Term Sequelae in the Hypothalamic Endocrine System. Horm Res Paediatr. 2021; 94(1-2): 9-17.
- DiCaprio MR, Roberts TT. Diagnosis and Management of Langerhans Cell Histiocytosis. Journal of the American Academy of Orthopaedic Surgeons. 2014; 22(10): 643-652.
- Abla O, Rollins B, Ladisch S. Langerhans cell histiocytosis: Progress and controversies. Br J Haematol. 2019; 187(5): 559-562.
- Medina MÁ, Meyer W, Echeverri C, Builes N. Histiocitosis de células de Langerhans: Reporte de caso y revisión de la literatura. Biomédica. 2021; 41(3): 396-402.
- Bagnasco F, Zimmermann SY, Egeler RM, Nanduri VR, Cammarata B, Donadieu J, et al. Langerhans cell histiocytosis and associated malignancies: A retrospective analysis of 270 patients. Eur J Cancer. 2022; 172: 138-145.
- Rodriguez‐Galindo C. Clinical features and treatment of Langerhans cell histiocytosis. Acta Paediatr. 2021; 10(11): 2892-2902.